刘小泽写于19.8.8-第三单元前四讲:学习scRNAseq这个R包
笔记目的:根据生信技能树的单细胞转录组课程探索smart-seq2技术相关的分析技术
课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=53
前言
内容在:https://github.com/jmzeng1314/scRNA_smart_seq2/blob/master/scRNA/study_scRNAseq.html
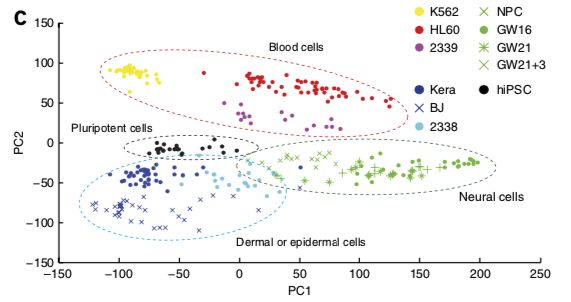
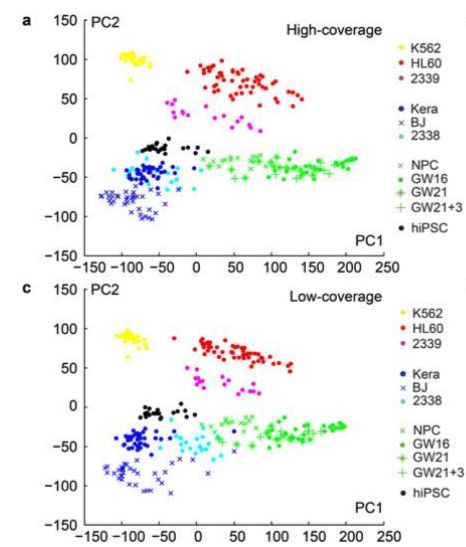
要使用scRNAseq这个R包,首先要对它进行了解,包中内置了Pollen et al. 2014 的数据集(https://www.nature.com/articles/nbt.2967),到19年8月为止,已经有446引用量了。只不过原文完整的数据是 23730 features, 301 samples,这个包中只选取了4种细胞类型:pluripotent stem cells 分化而成的 neural progenitor cells (NPC,神经前体细胞) ,还有 GW16(radial glia,放射状胶质细胞) 、GW21(newborn neuron,新生儿神经元) 、GW21+3(maturing neuron,成熟神经元) ,它们的关系如下图(NPC和其他三类存在较大差别):

要想知道数据怎么处理的,可以看:https://hemberg-lab.github.io/scRNA.seq.datasets/human/tissues/
简单看看文章怎么说
文章题目是:Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex
粗略看一下,不要求全文通读。作者利用低覆盖度单细胞转录组测序,揭示了大脑皮层发育过程的细胞异质性和激活的信号通路
研究背景
大规模的单细胞表达谱测序具有鉴定罕见细胞类型和发育关系的潜力,但需要有效地细胞捕获和mRNA测序方法。目前使用cell barcoding技术可以实现极低深度的并行测序,但是这种低深度测序有没有什么弊端还不知道。
文章使用了11个细胞群体的301个细胞进行了低深度测序(大约每个细胞测50000条reads),发现这种方法也可以和高深度一样进行细胞类型鉴定和biomarker鉴定。
材料方法
分为高深度测序和低深度测序,总共301个细胞
高深度:~8.9 × 106 reads per cell
低深度:~2.7 x 105 reads per cell数据在: http://www.ncbi.nlm.nih.gov/sra/?term=SRP041736
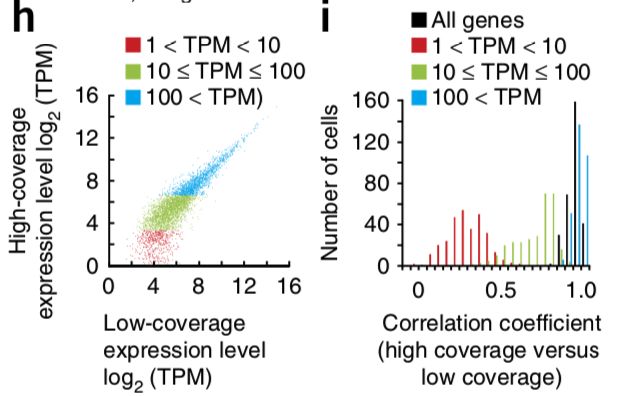
结论一:低覆盖度和高覆盖度的测序结果没太大区别
论点一:利用低覆盖度得到的spike-in含量和它们已知的浓度相关性很高(r = 0,968),当每次反应有32拷贝以上的spike-in时,所有的spike-in在所有样本中都能检测出,并且差异不大
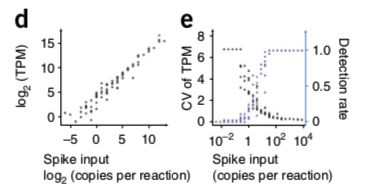
论点二:利用高覆盖度检测到的大部分基因,在低覆盖度方法中也能检测到
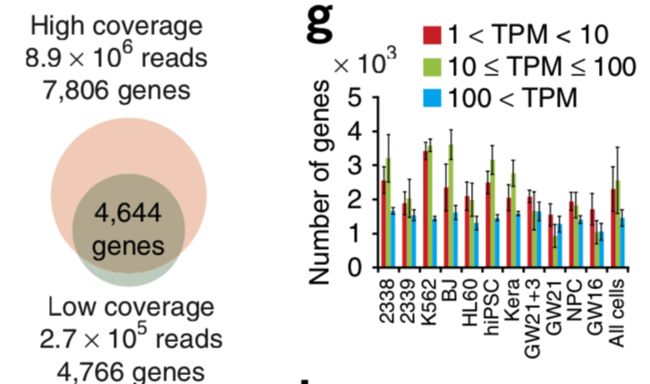
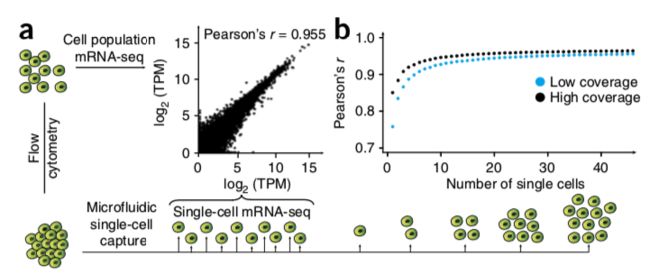
论点三:只在高覆盖度方法中检测到的基因,98%不是高表达(TPM>100),大多数(63%)是低表达(1 论点四:对于不同来源的的301个细胞,低覆盖度和高覆盖度得到的不同基因表达量估值的平均相关性为0.91 疑问一:但是呢,利用低覆盖度方法测序得到的低表达量基因(1 论点五: 虽然有局限,但是就取样技术来说,使用微流控(Microfluidic)获得的10个K562细胞和使用流式细胞术(flow cytometry)得到的一大群细胞,它们得到的表达量相关性很强(r = 0.955) 论点一: 先利用PCA看看细胞分群的效果 论点二:看看不同分组的基因表达情况 论点三: 低覆盖度和高覆盖度得到的细胞分布很相似,而且PCA得到的贡献最大的500个基因中,有78%是在低覆盖度和高覆盖度中都存在的 第一次见不会怎么办?看帮助文档和Bioconductor相关包的教程 也就是说,虽然原文总共做了301个样本,但这里一共有130个样本文库(高覆盖度、低覆盖度各65个) 具体数据的处理可以看这个R包的Bioconductor详细文档:https://bioconductor.org/packages/release/data/experiment/vignettes/scRNAseq/inst/doc/scRNAseq.html 其中介绍了另外两个数据集的来历,浏览一下: 这个包中的数据都是以 使用 The SummarizedExperiment class is a matrix-like container where rows represent features of interest (e.g. genes, transcripts, exons, etc...) and columns represent samples (with sample data summarized as a DataFrame). 以第一个数据 下面就是对这个对象的探索了: 看完表达矩阵,少不了的是样本的注释信息,这些就存放在了: 想法是:对每个QC指标都做个箱线图,这些指标会以向量的形式保存,然后一个一个循环操作,那么会向量循环就用 然后利用表型信息对样本进行过滤: 从下面这个箱线图中也可以看到,很少有基因在99个过滤后的细胞中都有表达,大部分基因还是在部分细胞中表达量为0 (这也是单细胞一个很特殊的现象:dropout情况,意思就是真实情况下基因是有表达量的,但技术问题没有检测到) 需要加上分组的信息: 小结:NPC跟另外的GW细胞群可以区分的很好,但是GW本身的3个小群体并没有那么好的区分度。后来简单选取mad前500的基因重新计算,也没有改善;另外可以看到每个细胞测了两次(图中对角线中有红色和橙色,表示两次不同深度) 算法很多,比如:主成分分析PCA、多维缩放(MDS)、线性判别分析(LDA)、等度量映射(Isomap)、局部线性嵌入(LLE)、t-SNE、Deep Autoencoder Networks PCA函数返回的结果如下:现在细胞在行,基因在列,所以细胞是 小结:NPC跟另外的GW细胞群可以区分的很好,但是GW本身的3个小群体并没有那么好的区分度 这里先选取PCA后的主成分,然进行tSNE;其实还可以选取变化高的基因,显著差异基因等等 降维和聚类不是一回事,各有各的算法、参数,比如降维我们常用PCA、tsne,聚类就有kmeans、dbscan 进行一个比较: 具体的算法可以不用了解太深 这里需要注意:如果提示找不到 同样的,如果函数 当然,可以挑选一些marker基因进行作图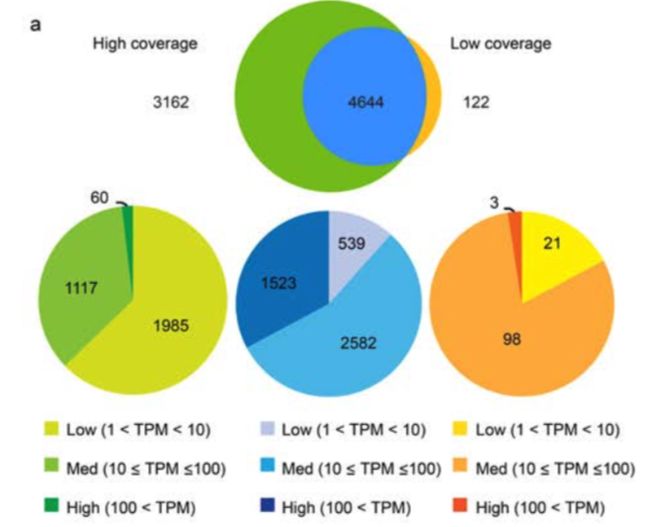
结论二: 低覆盖度测序也能区分不同细胞类型
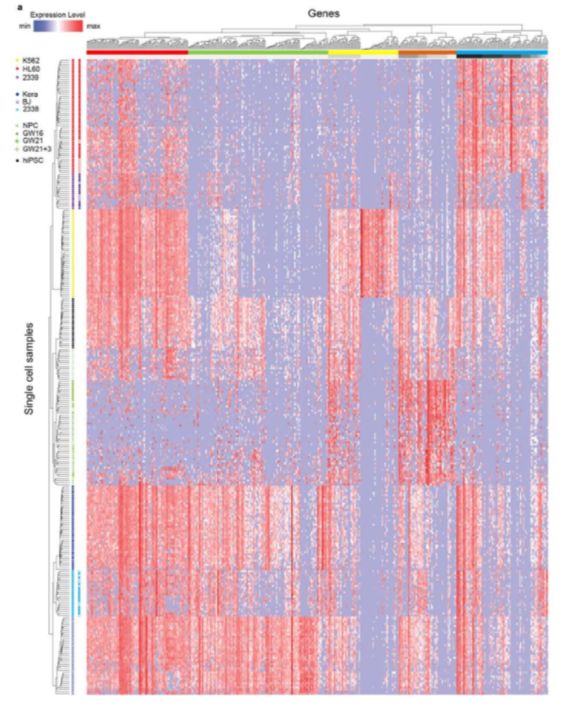
然后主要看这个包中的数据
# 先看帮助文档
library(scRNAseq)
?scRNAseq
# 其中包含了三种数据集:fluidigm、th2、allen,我们用到的是第一个fluidigm
# The dataset fluidigm contains 65 cells from Pollen et al. (2014), each sequenced at high and low coverage (SRA: SRP041736).
th2 contains 96 T helper cells from (Mahata et al. 2014) (ArrayExpress: E-MTAB-2512).allen contains 379 cells from the mouse visual cortex. This is a subset of the data published in (Tasic et al. 2016) (SRA: SRP061902).还介绍了数据的预处理:

直接看看数据应该怎么获取?
SummarizedExperiment对象形式存放的,那么什么是SummarizedExperiment对象?
?assay得到的帮助结果:fluidigm为例进行读取:library(scRNAseq)
data(fluidigm)
> fluidigm
class: SummarizedExperiment
dim: 26255 130
metadata(3): sample_info clusters which_qc
assays(4): tophat_counts cufflinks_fpkm rsem_counts rsem_tpm
rownames(26255): A1BG A1BG-AS1 ... ZZEF1 ZZZ3
rowData names(0):
colnames(130): SRR1275356 SRR1274090 ... SRR1275366 SRR1275261
colData names(28): NREADS NALIGNED ... Cluster1 Cluster2
# 例如要提取基因表达量的信息,就用assay函数(注意上面assays那一行,其中包含了4个结果:tophat_counts、 cufflinks_fpkm、rsem_counts、rsem_tpm)
names(assays(fluidigm))
## [1] "tophat_counts" "cufflinks_fpkm" "rsem_counts" "rsem_tpm"
# 默认访问第一个,也就是原始的表达量tophat_counts
head(assay(fluidigm)[,1:3])
## SRR1275356 SRR1274090 SRR1275251
## A1BG 0 0 0
## A1BG-AS1 0 0 0
## A1CF 0 0 0
## A2M 0 0 0
## A2M-AS1 0 0 0
## A2ML1 0 0 0
# 如果要得到RPKM值,可以使用assay:
head(assay(fluidigm, 2)[,1:3])
## SRR1275356 SRR1274090 SRR1275251
## A1BG 0 0.0000000 0
## A1BG-AS1 0 0.3256690 0
## A1CF 0 0.0687904 0
## A2M 0 0.0000000 0
## A2M-AS1 0 0.0000000 0
## A2ML1 0 1.3115300 0
# 或者使用assays
head(assays(fluidigm)$cufflinks_fpkm)
colData中# 包含了太多的信息,如果你直接使用colData(fluidigm),会得到眼花缭乱的结果
# 于是可以先大体看看有哪些类
names(metadata(fluidigm))
## [1] "sample_info" "clusters" "which_qc"
# 然后假如我们想看QC相关的信息(也是最常用的)
metadata(fluidigm)$which_qc
## [1] "NREADS" "NALIGNED"
## [3] "RALIGN" "TOTAL_DUP"
## [5] "PRIMER" "INSERT_SZ"
## [7] "INSERT_SZ_STD" "COMPLEXITY"
## [9] "NDUPR" "PCT_RIBOSOMAL_BASES"
## [11] "PCT_CODING_BASES" "PCT_UTR_BASES"
## [13] "PCT_INTRONIC_BASES" "PCT_INTERGENIC_BASES"
## [15] "PCT_MRNA_BASES" "MEDIAN_CV_COVERAGE"
## [17] "MEDIAN_5PRIME_BIAS" "MEDIAN_3PRIME_BIAS"
## [19] "MEDIAN_5PRIME_TO_3PRIME_BIAS"
# 因此我们想获得样本QC信息,就可以
sample_qc <- as.data.frame(colData(fluidigm)[metadata(fluidigm)$which_qc])
探索完,开始基本操作
# 我们要对RSEM得到的count值进行操作,之所以使用floor函数,是因为这个RSEM矩阵存在小数点。猜测:因为RSEM计算表达量是考虑了reads比对到不同基因的情况,这样的话就不能直接判断这个reads到底属于哪个基因,于是就用带小数的expected count(也就是真实值)表示。其实我们使用的时候,是需要变成整数(raw count)的,于是简单使用了floor向下取整
mtx <- floor(assay(fluidigm,3))
> mtx[1:3,1:3]
SRR1275356 SRR1274090 SRR1275251
A1BG 0 0 0
A1BG-AS1 0 0 0
A1CF 0 0 0
> dim(mtx)
[1] 26255 130
看表型信息并过滤
lapply# 目前QC指标都存在:colnames(sample_qc)中,一共19个;使用更容易调参数的ggboxplot
library(ggpubr)
box <- lapply(colnames(sample_qc),function(i) {
dat <- sample_qc[,i,drop=F]
dat$all_cells="all_cells"
ggboxplot(dat,x=dat[,2],y=i,
xlab=F,add = "jitter")
})
plot_grid(plotlist=box, ncol=5 )
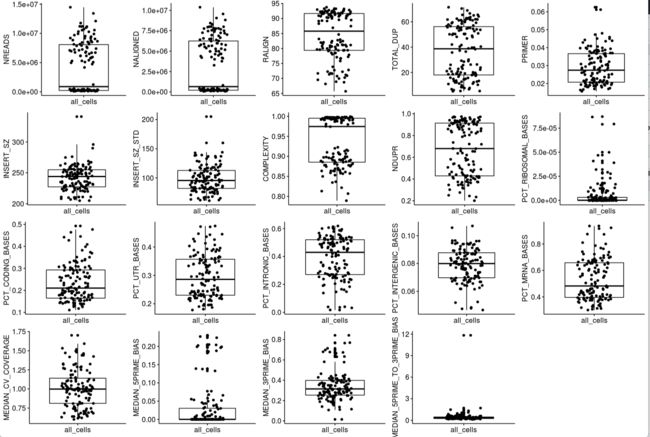
# 从QC数据中挑选一些指标,作为过滤条件
choose_anno <- colnames(sample_qc[,c(1:9,11:16,18,19)])
# 下面就是将一个个的QC条件进行细胞的过滤,如果细胞满足设定的QC过滤条件,就为1;否则为0;并且用cbind按列组合在一起
filter <- lapply(choose_anno,function(i) {
# 写循环时可以先用一个值作为测试:例如 i=choose_anno[1]
dat <- sample_qc[,i]
dat <- abs(log10(dat))
fivenum(dat)
(up <- mean(dat)+2*sd(dat))
(down <- mean(dat)- 2*sd(dat) )
valid <- ifelse(dat > down & dat < up, 1,0 )
})
filter <- do.call(cbind,filter)
# 得到了列为QC,行为细胞的过滤结果,那么就将QC条件全部为1的细胞挑出来(也就是找全部为1的行)
choosed_cells <- apply(filter,1,function(x) all(x==1))
# 进行对比:原来的细胞
> table(colData(fluidigm)$Biological_Condition)
GW16 GW21 GW21+3 NPC
52 16 32 30
# 过滤后的细胞
> table(colData(fluidigm)[choosed_cells,]$Biological_Condition)
GW16 GW21 GW21+3 NPC
36 11 23 29
# 将表达矩阵进行过滤
mtx <- mtx[,choosed_cells]
> dim(mtx)
[1] 26255 99
看基因表达信息
> fivenum(apply(mtx,1,function(x) sum(x>0) ))
A1CF OR8G1 LINC01003 MRPS36 YWHAZ
0 0 4 26 99
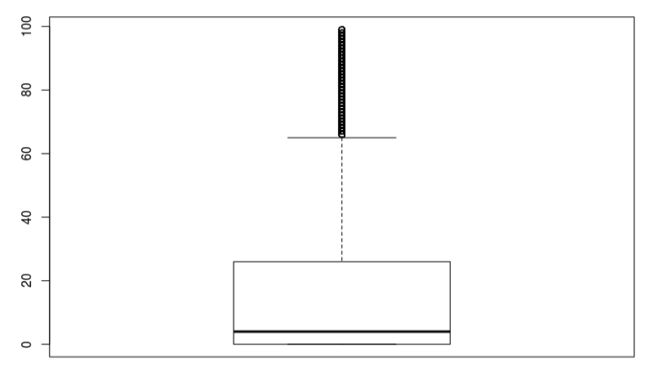
# 看到至少有25%的基因表达量为0.那么具体有多少个呢?可以看看:
choosed_genes=apply(mtx,1,function(x) sum(x>0) )>0
> table(choosed_genes)
FALSE TRUE
9496 16759
# 看到有9000多个基因在所有细胞中都没有表达量(可能原因:原文分析的确实是2w多个基因,但他是在300多个细胞中分析的,我们这个包里过滤后只剩下99个细胞,所以存在很多基因不在这部分细胞中表达,因此需要去掉)
boxplot(apply(mtx,1,function(x) sum(x>0) ))
# 最后根据基因表达量对矩阵进行过滤
mtx <- mtx[choosed_genes,]
看细胞间基因表达量相关性
# 都要基于CPM标准化数值,并做一个备份
mtx <- log2(edgeR::cpm(mtx) + 1)
mtx[1:4, 1:4]
mtx_back <- mtx
# 对相关性进行初步的可视化
exprSet <- mtx_back
> dim(exprSet)
[1] 16759 99
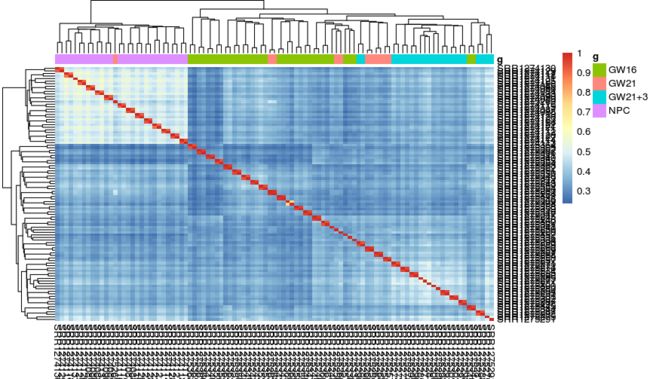
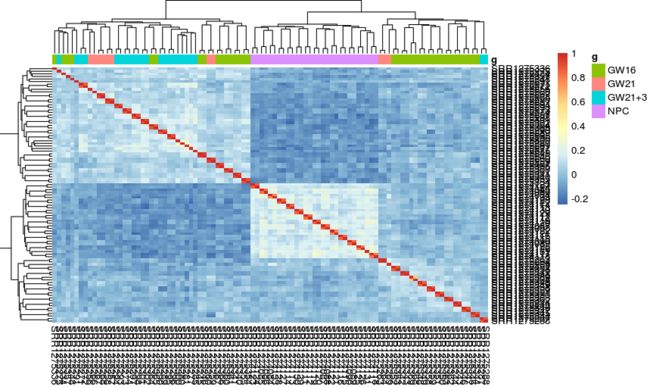
pheatmap::pheatmap(cor(exprSet))
# 注意:cor函数计算的是列与列间的相关系数

# 使用细胞过滤后的分组信息(GW16:36 GW21:11 GW21+3:23 NPC:29)
group_list <- colData(fluidigm)[choosed_cells,]$Biological_Condition
tmp <- data.frame(g = group_list)
rownames(tmp) <- colnames(exprSet)
# 组内相似性高于组间,并且看到NPC组和其他组差异更大
pheatmap::pheatmap(cor(exprSet), annotation_col = tmp)
好,接着设置阈值对表达矩阵过滤
# 这一次设置阈值为5,表示至少要满足基因在5个细胞中的表达量都大于1
exprSet = exprSet[apply(exprSet, 1, function(x) sum(x > 1) > 5), ]
> dim(exprSet)
[1] 11337 99
过滤完,按照mad统计方法取前500个表达量变化最大的基因
# 绝对中位差来估计方差,先计算出数据与它们的中位数之间的偏差,然后这些偏差的绝对值的中位数就是mad
exprSet <- exprSet[names(sort(apply(exprSet, 1, mad), decreasing = T)[1:500]), ]
> dim(exprSet)
[1] 500 99
# 对组间差异最大的基因再进行相关性分析
M <-cor(log2(exprSet + 1))
tmp <- data.frame(g = group_list)
rownames(tmp) <- colnames(M)
pheatmap::pheatmap(M, annotation_col = tmp)
表达矩阵简单的层次聚类
mtx <- mtx_back
hc <- hclust(dist(t(mtx))) # dist以行为输入
plot(hc,labels = FALSE)

clus <- cutree(hc, 4) #对hclust()函数的聚类结果进行剪枝,即选择输出指定类别数的系谱聚类结果。
group_list <- as.factor(clus) ##转换为因子属性
> table(group_list) ##统计频数
group_list
1 2 3 4
29 25 39 6
filtered_anno <- colData(fluidigm)[choosed_cells,]$Biological_Condition
> table(group_list,filtered_anno)
filtered_anno
group_list GW16 GW21 GW21+3 NPC
1 0 0 0 29
2 20 3 2 0
3 15 8 16 0
4 1 0 5 0
# 结果看到:NPC、GW21+3这两个利用普通的层次聚类还是可以区分开,但是GW16、GW21就不太能区分了
最常规的PCA降维结果
以下会采用PCA 和 t-SNEmtx <- mtx_back
mtx <- t(mtx) # PCA也是对行操作:需要先转置一下,让行为样本
mtx <- as.data.frame(mtx)
plate <- filtered_anno # 这里定义分组信息
mtx <- cbind(mtx, plate) # 添加分组信息
> mtx[1:4, 1:4]
A1BG A1BG-AS1 A2M A2M-AS1
SRR1274090 0 0 0.000000 0
SRR1275287 0 0 4.216768 0
SRR1275364 0 0 0.000000 0
SRR1275269 0 0 3.552694 0
> table(mtx$plate)
GW16 GW21 GW21+3 NPC
36 11 23 29
# 进行PCA操作(实际是降维,映射到二维坐标)
mtx.pca <- PCA(mtx[, -ncol(mtx)], graph = FALSE)
> head(mtx.pca$var$coord) ## 每个主成分的基因重要性占比
Dim.1 Dim.2 Dim.3 Dim.4 Dim.5
A1BG 0.19046450 0.09601240 -0.17840553 -0.001507970 -0.0006057691
A1BG-AS1 -0.02510451 0.29821319 0.03571804 0.020001929 -0.0105727109
A2M 0.03403042 0.25458727 0.24264958 0.228512329 0.5414019044
A2M-AS1 0.23140893 0.02900348 -0.07952678 0.356461354 0.1283450099
A2ML1 -0.15776536 0.13831288 0.10065788 0.004060288 -0.0353422367
A2MP1 -0.04068586 -0.05584736 -0.02857416 0.018287992 0.0069603680
> head(mtx.pca$ind$coord) ## 每个细胞的前5个主成分取值。
Dim.1 Dim.2 Dim.3 Dim.4 Dim.5
SRR1274090 40.251912 -13.231641 -12.358891 -20.038100 -12.704947
SRR1275287 1.196637 15.386256 30.566235 14.262858 -4.852418
SRR1275364 -34.731051 -14.782146 -7.716928 7.046918 1.951473
SRR1275269 21.760471 3.307309 17.985263 -18.382512 9.270646
SRR1275263 -3.313968 -15.856721 8.929275 -36.358830 20.275875
SRR1274117 59.378486 16.453551 -5.098901 56.245455 19.257598
ind,基因是var
# 进行PCA可视化
fviz_pca_ind(
mtx.pca,
#repel =T,
geom.ind = "point",
# show points only (nbut not "text")
col.ind = mtx$plate,
# color by groups
#palette = c("#00AFBB", "#E7B800"),
addEllipses = TRUE,
# Concentration ellipses
legend.title = "Groups"
)

进阶一点的tSNE降维
# 选取前面PCA分析的5个主成分。
tsne_mtx <- mtx.pca$ind$coord
# Set a seed if you want reproducible results
set.seed(42)
library(Rtsne)
# 如果使用原始表达矩阵进行 tSNE耗时会很久
# 如果出现Remove duplicates before running TSNE 则check_duplicated = FALSE
# tsne_out <- Rtsne(dat_matrix,pca=FALSE,perplexity=30,theta=0.0, check_duplicates = FALSE)
# Run TSNE
tsne_out <- Rtsne(tsne_mtx,perplexity=10)
plate <- filtered_anno # 这里定义分组信息
plot(tsne_out$Y,col= rainbow(4)[as.numeric(as.factor(plate))], pch=19)
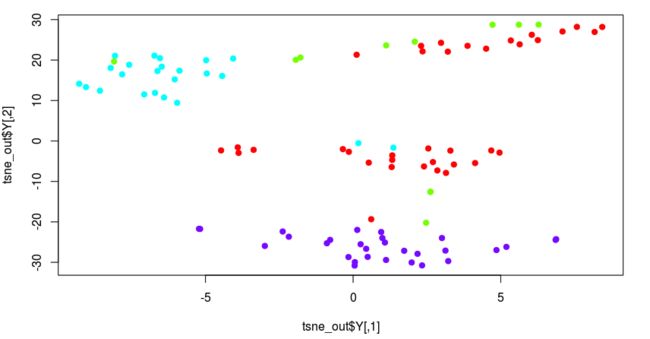
降维后呢?
> # 前面我们的层次聚类是针对全部表达矩阵,tsne选取前面PCA分析的5个主成分,因此为了节省计算量,选取tsne_out$Y这个结果
> head(tsne_out$Y)
[,1] [,2]
[1,] 4.855236 -26.9704714
[2,] 0.179925 -0.5475169
[3,] 6.256713 24.9241040
[4,] 2.471635 -20.2250523
[5,] 2.615960 -12.6056267
[6,] -2.384375 -22.3821087
opt_tsne=tsne_out$Y
> table(kmeans(opt_tsne,centers = 4)$clust)
1 2 3 4
31 24 24 20
plot(opt_tsne, col=kmeans(opt_tsne,centers = 4)$clust, pch=19, xlab="tSNE dim 1", ylab="tSNE dim 2")

# 换一种dbscan
library(dbscan)
plot(opt_tsne, col=dbscan(opt_tsne,eps=3.1)$cluster, pch=19, xlab="tSNE dim 1", ylab="tSNE dim 2")
> table(dbscan(opt_tsne,eps=3.1)$cluster)
0 1 2 3 4
2 22 38 31 6
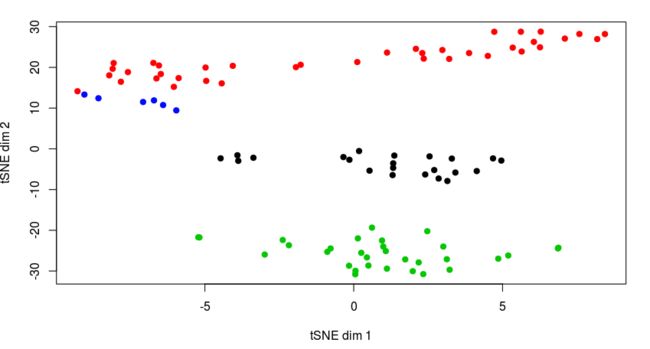
# 比较两个聚类算法区别
> table(kmeans(opt_tsne,centers = 4)$clust,dbscan(opt_tsne,eps=3.1)$cluster)
0 1 2 3 4
1 0 0 38 0 6
2 0 0 0 16 0
3 0 0 0 15 0
4 2 22 0 0 0
下面使用M3Drop包处理单细胞数据
第一步 构建对象
## 重新加载数据
rm(list=ls())
data(fluidigm)
# names(assays(fluidigm))
counts <- floor(assay(fluidigm, 3))
dim(counts)
## 过滤
sample_qc <- as.data.frame(colData(fluidigm)[metadata(fluidigm)$which_qc])
choose_anno <- colnames(sample_qc[,c(1:9,11:16,18,19)])
filter <- lapply(choose_anno,function(i) {
# i=choose_anno[1]
dat <- sample_qc[,i]
dat <- abs(log10(dat))
fivenum(dat)
(up <- mean(dat)+2*sd(dat))
(down <- mean(dat)- 2*sd(dat) )
valid <- ifelse(dat > down & dat < up, 1,0 )
})
filter <- do.call(cbind,filter)
choosed_cells <- apply(filter,1,function(x) all(x==1))
counts <- counts[,choosed_cells]
## 开始M3Drop分析
library(M3Drop)
Normalized_data <- M3DropCleanData(counts,
labels = colData(fluidigm)[choosed_cells,]$Biological_Condition,
is.counts=TRUE, min_detected_genes=2000)
> dim(Normalized_data$data)
[1] 13405 97
## 检查
> str(Normalized_data) #只返回了一个list,而不是S4对象
List of 2
$ data : num [1:13405, 1:97] 0 0 0 0 0 0 0 0 0 0 ...
..- attr(*, "dimnames")=List of 2
.. ..$ : chr [1:13405] "A1BG" "A2M" "A2ML1" "AAAS" ...
.. ..$ : chr [1:97] "SRR1274090" "SRR1275287" "SRR1275364" "SRR1275269" ...
$ labels: chr [1:97] "NPC" "GW21+3" "GW16" "GW21" ...
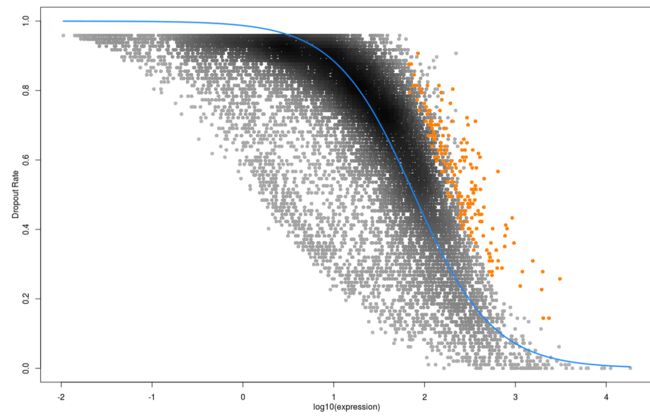
第二步 表达矩阵检验--Michaelis-Menten算法
fits <- M3DropDropoutModels(Normalized_data$data)

## 查看检验的结果
## Sum absolute residuals
data.frame(MM=fits$MMFit$SAr, Logistic=fits$LogiFit$SAr,
DoubleExpo=fits$ExpoFit$SAr)
# MM Logistic DoubleExpo
1 1651 1646 4033
## Sum absolute residuals
data.frame(MM=fits$MMFit$SAr, Logistic=fits$LogiFit$SAr,
DoubleExpo=fits$ExpoFit$SAr)
# MM Logistic DoubleExpo
1 403 345 1962
第三步 找差异基因
M3DropDifferentialExpression这个函数,那么可能由于安装的M3Drop包版本是旧版,新版对应的函数是:M3DropFeatureSelectionDE_genes <- M3DropFeatureSelection(Normalized_data$data,
mt_method="fdr", mt_threshold=0.01)
> dim(DE_genes)
[1] 182 4
> head(DE_genes)
Gene effect.size p.value q.value
IGFBPL1 IGFBPL1 13.94805 1.790836e-21 4.801233e-18
DLK1 DLK1 11.66363 1.104816e-22 4.936685e-19
BCAT1 BCAT1 10.97600 6.335191e-31 8.492323e-27
CA12 CA12 10.94265 1.020863e-08 3.110152e-06
ZNF30 ZNF30 10.78263 2.503772e-06 3.813985e-04
FOS FOS 10.01977 1.389763e-12 9.314889e-10
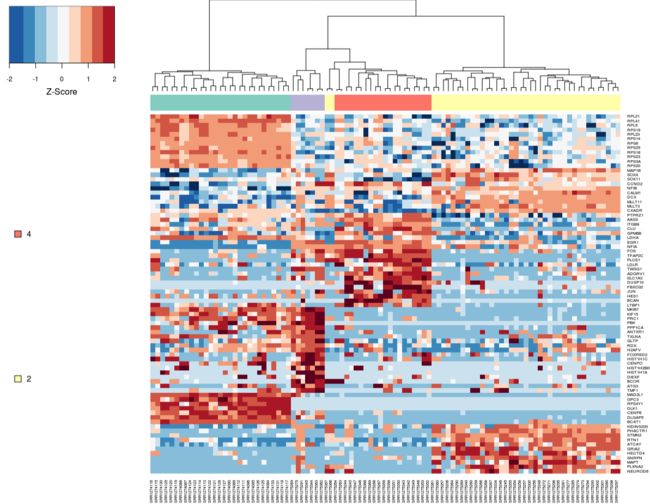
第四步 差异基因画热图
# 就是看一下差异基因在不同细胞类型的表达分布
par(mar=c(1,1,1,1))
heat_out <- M3DropExpressionHeatmap(DE_genes$Gene, Normalized_data$data,
cell_labels = Normalized_data$labels)
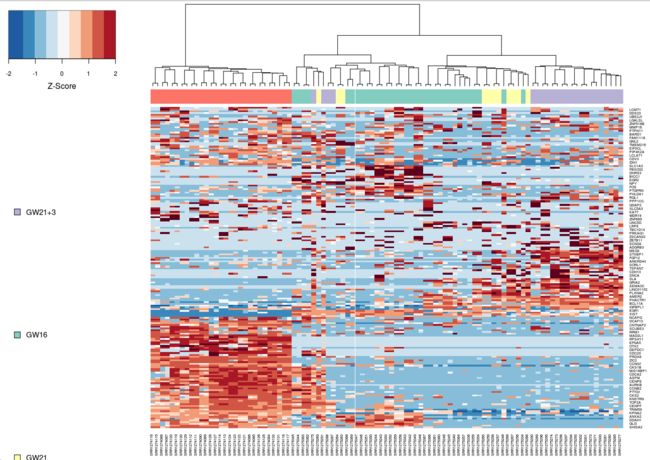
第五步 重新聚类,找marker
M3DropGetHeatmapCellClusters找不到,就替换成M3DropGetHeatmapClusters,因为作者时刻在改函数名字,有困难找:https://bioconductor.org/packages/release/bioc/vignettes/M3Drop/inst/doc/M3Drop_Vignette.R# 重新聚类
cell_populations <- M3DropGetHeatmapClusters(heat_out, k=4)
# 重聚类得到的和自带的表型数据比较
> table(cell_populations,Normalized_data$labels)
cell_populations GW16 GW21 GW21+3 NPC
1 0 0 0 29
2 14 8 19 0
3 4 1 2 0
4 16 2 2 0
# 找marker
library("ROCR")
marker_genes <- M3DropGetMarkers(Normalized_data$data, cell_populations)
第六步 可以看每个分类的marker基因
# 比如想看新得到的第4组的marker基因
> head(marker_genes[marker_genes$Group==4,],10)
AUC Group pval
ADGRV1 0.9707792 4 1.831217e-11
TFAP2C 0.9451299 4 1.885637e-11
EGR1 0.9409091 4 1.159852e-09
PLCE1 0.9233766 4 7.676760e-11
FOS 0.9058442 4 1.806229e-08
SLC1A3 0.9048701 4 1.542660e-09
AASS 0.9032468 4 3.136961e-08
ITGB8 0.8886364 4 8.823909e-08
BCAN 0.8844156 4 1.513264e-12
NFIA 0.8831169 4 7.724889e-08
# 或者想知道某个marker基因的分配位置
> marker_genes[rownames(marker_genes)=="FOS",]
AUC Group pval
FOS 0.9058442 4 1.806229e-08
# 挑选的代码
choosed_marker_genes=as.character(unlist(lapply(split(marker_genes,marker_genes$Group), function(x) (rownames(head(x,20))))))
# 看不懂?没关系,可以拆解~
# 其中最核心的是split(marker_genes,marker_genes$Group),它返回什么东西,用str()查看就对了
> str(split(marker_genes,marker_genes$Group))
List of 4
$ 1:'data.frame': 5370 obs. of 3 variables:
..$ AUC : num [1:5370] 1 0.998 0.998 0.997 0.993 ...
..$ Group: chr [1:5370] "1" "1" "1" "1" ...
..$ pval : num [1:5370] 8.68e-22 1.20e-17 1.04e-14 1.18e-14 1.81e-14 ...
$ 2:'data.frame': 1844 obs. of 3 variables:
..$ AUC : num [1:1844] 0.99 0.936 0.933 0.927 0.927 ...
..$ Group: chr [1:1844] "2" "2" "2" "2" ...
# 还有两段省略............
# 不难看出,它的作用是根据分组信息(marker_genes$Group),将marker_genes分成了4个部分,共同组成一个列表。现在我们可以最粗略地选出每个分组中的前20基因(当然,后续可以自定义,根据AUC和P值来挑选)
# 对列表循环用lapply,要做的事情就是挑出前20=》rownames(head(x,20))
# 接下来作图就容易了
par(mar=c(1,1,1,1))
heat_out <- M3DropExpressionHeatmap(choosed_marker_genes, Normalized_data$data, cell_labels = cell_populations)