00 写在前面
仅针对人类WGS或WES数据,供参考。
时间管理某一点:能自动化的工作尽量自动化,不要时间用在毫无意义的重复上。脑袋好比肌肉,需要不断提高负重的锻炼才能达到“有效锻炼”。
01 软件准备
这里主要使用GATK的best practice进行数据处理,GATK一直在更新维护,建议下载最新版,这里用的是GenomeAnalysisTK-3.8-0-ge9d806836。参考链接如下:https://software.broadinstitute.org/gatk/
GATK的维护做得很棒,在使用过程中我遇到的所有问题(各种报错信息)都能在官网搜索到解决方案。擅于搜索,也是学习。

流程化操作主要包括以下三部分:
- 测序数据评估及质控:评估测序质量(fastqc,multiqc),去除接头(cutadapt),去除reads两端低质量碱基(fastx_toolkit)
- SNV calling:序列比对(bwa),索引建立及统计比对情况(samtools),建立索引( picard),SNV检测(gatk)
- SNV 过滤:gatk,数据库。
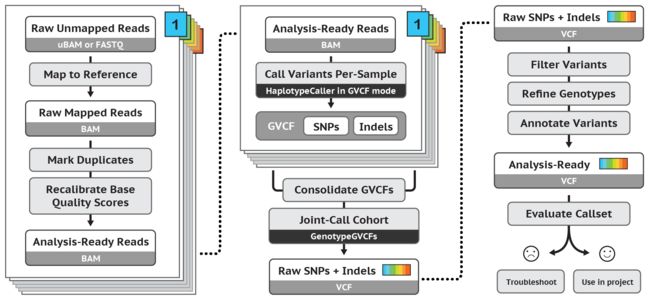 流程
流程
优先使用conda安装软件,网络OK的情况下,方便快捷,可以很好地解决依赖问题;不行的话,下载安装至biosoft/下
conda install fastqc
conda install multiqc
···
picard="/biosoft/picard-tools-1.124/picard.jar" #添加到.bash_profile
java -jar $picard -h
GATK=/biosoft/GenomeAnalysisTK-3.8-0-ge9d806836/GenomeAnalysisTK.jar #添加到.bash_profile
java -jar $GATK -h
02 数据库准备
- 下载参考基因组
常用版本为hg19和hg38,建议使用hg38(最新版)。网速不给力时,用Aspera下载,非常快。
下载链接:http://hgdownload.cse.ucsc.edu/downloads.html
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/
#下载某条染色体:如chr22
mkdir DB/ && cd DB/
DB="~/../DB/"
wget --timestamping 'ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/chr22.fa.gz'
gzip -d chr22.fq.gz
#下载hg19
wget --timestamping 'ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/*';
md5sum filename #下载的文件较多时,校验md5以确定文件是完整的
#合并为hg19.fa
gunzip chr[1-22].gz
gunzip chrX.gz chrY.gz chrM.gz
for i in $(seq 1 22) X Y M; do cat chr${i}.fa >>hg19.fa; done
rm -fr chr*.fasta
#下载hg38
wget --timestamping 'ftp://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/hg38.fa.gz';
- 下载数据库
从GATK resource bundle下载dbSNP,1000G相关数据,用于GATK best practice过程中对变异进行校正。
GATK_FTP=ftp://[email protected]/bundle/hg19
wget $GATK_FTP/dbsnp_138.hg19.vcf.gz
wget $GATK_FTP/1000G_phase1.indels.hg19.sites.vcf.gz
wget $GATK_FTP/Mills_and_1000G_gold_standard.indels.hg19.sites.vcf.gz
03 测序数据质量评估及质控
03-1 质量评估Fastqc, multiqc
一般公司提交的是初步质控处理过的clean data,最好把raw data一并要过来。一般公司会对数据进行基本的质控,但我们还要再确认一次,为什么呢?个人遇到的情况包括以下几种:
- 部分样本的接头可能去得不完全
- 此外,为了数据量好看(更大),一般质量过得去的情况下,公司不会对reads前后端测序质量较低的碱基进行剪切,需要自己根据情况进行处理
- 有的公司对不同样本处理不一致,每个样本的clean data中reads长度可能不一致,有些后续分析是要求不同样本的reads一致
以上情况发生时,就需要自己对数据进行(再次)质控了。
cd fqDIR/ #fq文件存放位置
mkdir qcDIR/ qcDIR_multiqc/ #qc结果存放位置; multiqc结果存放位置
gzip sample1_1.fq.gz #写Shell脚本批量操作
echo "Spend time:$SECONDS\n" #This line will tell u how long it takes to finish the job
fastqc sample1_1.fq -o qcDIR/ -t 6 -d qcDIR/ >>fastqc.r.log 2>>fastqc.e.log
#利用multiqc整合结果,方便批量查看
multiqc qcDIR/ -o qcDIR_multiqc/
结果如下(fastqc_per_base_sequence_quality_plot)所示

03-2 去除INDEX和接头
一般测序,每个样本有独特的INDEX,还有通用的接头。需要与公司确认好具体的建库方式,INDEX序列,两端接头序列(注意方向),不然一不小心切错了都不知道...
#[待检测]
cutadapt -a ADAPTER_FWD -A ADAPTER_REV --pair-filter=both -o sample1_cut_1.fq -p sample1_cut_2.fq sample1_1.fq sample1_2.fq
#注意更改INDEX序列,adapter方向
03-3 去除低质量碱基,过滤低质量reads
fastx_toolkit:http://hannonlab.cshl.edu/fastx_toolkit/commandline.html
03-4 再次质量评估
fastqc, multiqc
最后得到clean Data,用于后续分析,如SNV calling
对germline变异,现在最常用的是GATK的best practice流程。如果是癌症样本,GATK有对应的流程,也有其他的软件,如Varscan等,不在本文讨论范围内。
04 SNV calling
04-1 数据预处理: map, sort, mark duplicates, base recalibration
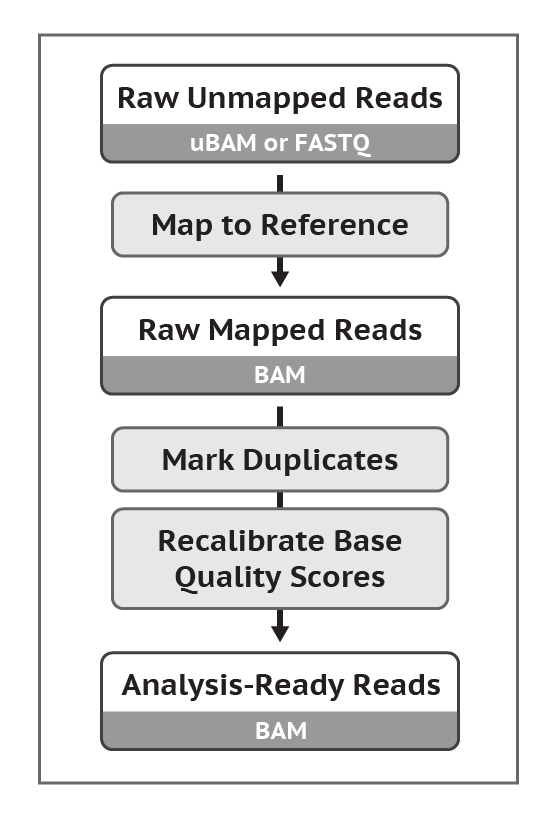
给参考基因组建立索引
#create index
samtools faidx hg19.fa
#creat dictionary, required by many processing and analysis tools.
java -jar $picard CreateSequenceDictionary\
R=hg19.fa\
O=hg19.dict #该文件可以看到每条染色体的命名,后面QBSR时指定染色体需用到
比对到参考基因组,生成SAM文件
bwa index -a bwtsw hg19.fa #*.fa >2G,用-a bwtsw;*.fa < 2G,用-a is (默认)
time bwa mem -M -t 10 -R '@RG\tID:HKV2KALXX.7\tSM:sample1\tPL:illumina\tLB:sample1'
$DB/hg19.fa sample1_1.fq sample1_2.fq >aligned_sample1.sam
#-M :Mark shorter split hits as secondary (essential for Picard compatibility)
#-t: number of threads
#-R:定义头文件,如果在此步骤不进行头文件定义,在后续GATK分析中还是需要重新增加头文件。具体信息可从样本的fq文件中获得。
#@RG: Read Group,必须要有,否则GATK无法进行calling;比对速度比不加@RG更快
##ID:Read group identifier, flowcell + lane name and number in Illunima data >>>> ID:FLOWCELL1.LANE1(每个flowcell的每个lane是unique的), EX. HKV2KALXX.7
#PL: platform>>> ILLUMINA
#SM: Sample >>>sample1
#LB: DNA preparation library identifier. MarkDuplicates uses the LB to determine which read groups might contain molecular duplicates, in case the same DNA library was sequenced on multiple lanes.
#PU:Platform Unit >>>HKV2KALXX.2 #太复杂,非必须。
转换成BAM文件,sort, mark duplicates
java -jar $picard SortSam \
INPUT=aligned_sample1.sam \
OUTPUT=sorted_sample1.bam \
SORT_ORDER=coordinate
java -jar $picard MarkDuplicates \
INPUT=sorted_sample1.bam \
OUTPUT=dedup_sample1.bam \
METRICS_FILE=metrics_sample1.txt
#just mark, not remove. most tools in GATK will ingore the marked reads;If desired, duplicates can be removed using the REMOVE_DUPLICATE and REMOVE_SEQUENCING_DUPLICATES options.
java -jar $picard BuildBamIndex \
INPUT=dedup_sample1.bam
#查看@RG
samtools view -H dedup_sample1.bam | grep '@RG'
如果同一样本(同一文库)多次上样测序,需要将多次测序结果合并后,再进行sort,mark。
#merge bam files(利用picard可以保留@RG; samtools合并时会丢掉@RG)
time java -jar $picard MergeSamFiles\
I=1.bam I=2.bam\
O=12.bam
Recalibrate base quality scores = run BQSR
校正测序错误和部分实验所致误差
#analyze patterns of covariation in the dataset
java -jar $GATK \
-T BaseRecalibrator \
-R $DB/hg19.fa \
-I dedup_sample1.bam \
-L chr22 \ # restrict analysis to only chromosome 22,名称参考前面hg19.dict文件
-knownSites $DB/dbsnp_138.hg19.vcf \
-knownSites $DB/Mills_and_1000G_gold_standard.indels.hg19.sites.vcf \
-knownSites $DB/1000G_phase1.indels.hg19.sites.vcf \
-o recal_data_sample1.table
#a second pass to analyze covariation remaining after recalibartion
java -jar $GATK \
-T BaseRecalibrator \
-R $DB/hg19.fa \
-I 12unRG_dedup.bam \
-L chr22 \
-knownSites $DB/dbsnp_138.hg19.vcf \
-knownSites $DB/Mills_and_1000G_gold_standard.indels.hg19.sites.vcf \
-knownSites $DB/1000G_phase1.indels.hg19.sites.vcf \
-BQSR recal_data_sample1.table
-o post_recal_data_sample1.table
#generate before/after plots
java -jar $GATK \
-T AnalyzeCovariates \
-R $DB/hg19.fa \
-L chr22 \
-before recal_data_sample1.table \
-after post_recal_data_sample1.table \
-plots recalibration_sample1_plots.pdf
#Apply the recalibration to your sequence data
java -jar $GATK \
-T PrintReads \
-R $DB/hg19.fa \
-I dedup_sample1.bam \
-L chr22 \
-BQSR recal_data_sample1.table \
-o recal_sample1.bam
04-2 检测SNV: calling, consolidate, filter(VQSR/hard filter)
Note: 2013以前HaplotypeCaller还没完全开发好,用UnifiedGenotyper较多;目前作者推荐使用HaplotypeCaller,在indel检测上更准确。
Call variants per-sample with HaplotypeCaller (in GVCF mode)
java -jar $GATK \
-T HaplotypeCaller \
-R $DB/hg19.fa \
-I dedup_sample1.bam \
-L targets.interval_list \ #使用exome数据时,可用该参数指定文件(exome targets);也可以直接指定某染色体,如chr22
--genotyping_mode DISCOVERY \ #默认参数;GENOTYPE_GIVEN_ALLELES,仅使用给定文件中的allele
-stand_call_conf 30 \ #minimum confidence threshold (phred-scaled) at which the program should emit variant sites as called; 低于该值则标记为“LowQual"
-ERC GVCF \
-o sample1.raw_variants.g.vcf
Note : targets.interval_list 格式 不同的格式对应不同后缀名
https://gatkforums.broadinstitute.org/gatk/discussion/1319/collected-faqs-about-interval-lists
- Picard-style .interval_list
必须有SAM-like header;
@HD VN:1.0 SO:coordinate
@SQ SN:1 LN:249250621 AS:GRCh37 UR:http://www.broadinstitute.org/ftp/pub/seq/references/Homo_sapiens_assembly19.fasta M5:1b22b98cdeb4a9304cb5d48026a85128 SP:Homo Sapiens
@SQ SN:2 LN:243199373 AS:GRCh37 UR:http://www.broadinstitute.org/ftp/pub/seq/references/Homo_sapiens_assembly19.fasta M5:a0d9851da00400dec1098a9255ac712e SP:Homo Sapiens
1 30366 30503 + target_1
1 69089 70010 + target_2
1 367657 368599 + target_3
1 621094 622036 + target_4
- GATK-style .list
: - - BED files with extension .bed
- VCF files
Consolidate GVCFs
Joint-Call Cohort:GenomicsDBImport GenotypeGVCFs**
合并多个样本的g.vcf文件,用于群体的Genotype;有这一步比分开做genotype更准确。如果有新增样本,可以在这一步加入。
#Consolidate GVCFs with `GenomicsDBImport` or `CombineGVCFs`**
#GenomicsDBImport:新方法,速度快,但目前一次只能处理一条染色体
gatk GenomicsDBImport \
-V gvcfs/sample1.g.vcf \
-V gvcfs/sample2.g.vcf \
-V gvcfs/sample3.g.vcf \ #样本较多,可以用-V gvcfs/*.g.vcf
--genomicsdb-workspace-path my_database \ #自动生成存放结果的文件夹
--intervals chr22 #目前一次只能处理一条染色体,或一个interval (2018);可以用脚本分别操作后,再用CombineVariants合并。
#joint genotyping
java -jar $GATK \
-T GenotypeGVCFs \
-R $DB/hg19.fa \
-V gendb://my_database \
-G StandardAnnotation -newQual \
-O raw_variants.vcf
#CombineGVCFs:旧方法,速度慢,但是可以一次全部合并(合并不同样本的文件)
java -jar $GATK \
-T CombineGVCFs \
-R $DB/hg19.fa \
-V sample1.g.vcf \
-V sample2.g.vcf \
-o combined.g.vcf
#样本较多时,按如下操作
find gvcf/ -name "*.g.vcf" >input.list
nohup java -Xmx20g -jar $GATK -T CombineGVCFs -R $DB/hg19.fa --variant input.list -o combined.g.vcf & #Xmx20g 使用20G内存
#joint genotyping
java -jar $GATK \
-T GenotypeGVCFs \
-R $DB/hg19.fa \
-V combined.g.vcf \
-G StandardAnnotation -newQual \
-o raw_variants.vcf
NOTE:"." in the filter field is not bad (it means that no filtering was applied).
FILTER field:
PASS if the variant passed all filters.
. no filtering has been applied to the records.
It is extremely important to apply appropriate filters before using a variant callset in downstream analysis. See our documentation on filtering variants for more information on this topic.
Filter Variants by Variant (Quality Score) Recalibration:VariantRecalibrator ApplyRecalibration
GATK检测变异较为lenient,以确保不会遗漏真实的变异,但因此也会有假阳性高的问题,因此还需要进一步的过滤。过滤方法有两种:
- VQSR:要求样本量大(至少30个WES或1个WGS、有足够的已有变异信息(比如人类或模式生物);否则请用hard filtering
- hard filtering: 利用vcf中
INFO中的参数进行过滤
VQSR: recalibrate variant quality scores and produce a callset filtered for the desired levels of sensitivity and specificity.(利用HapMap,OMIM,1000G,dbSNP数据对SNP进行校正;利用Mills, dbSNP对indel进行校正)
hard filtering 公司常用的过滤标准:
GATK_SNP_Filter=--filterExpression 'DP<10' --filterName LOW_DP --filterExpression 'QD<2.0' --filterName LOW_QD --filterExpression 'FS>60.0' --filterName HIGH_FS --filterExpression 'SOR>4.0' --filterName HIGH_SOR --filterExpression 'MQ<40.0' --filterName LOW_MQ --filterExpression 'MQRankSum<-12.5' --filterName LOW_MQRS --filterExpression 'ReadPosRankSum<-8.0' --filterName LOW_RPRS -disable_auto_index_creation_and_locking_when_reading_rods
GATK_INDEL_Filter=--filterExpression 'DP<10' --filterName LOW_DP --filterExpression 'QD<2.0' --filterName LOW_QD --filterExpression 'FS>200.0' --filterName HIGH_FS --filterExpression 'SOR>10.0' --filterName HIGH_SOR --filterExpression 'InbreedingCoeff<-0.8' --filterName LOW_INC --filterExpression 'ReadPosRankSum<-20.0' --filterName LOW_RPRS -disable_auto_index_creation_and_locking_when_reading_rods
下面介绍VQSR
- 下载数据:hapmap_3.3.hg19.vcf 1000G_omni2.5.hg19.vcf 1000G_phase1.snps.high_confidence.hg19.vcf dbsnp_138.hg19.vcf, Mills_and_1000G_gold_standard.indels.hg19.vcf
FileZile host: ftp.broadinstitute.org user:gsapubftp-anonymous (md5sum filename,确认文件完整性(md5))
wget ftp://[email protected]/bundle/hg19/1000G_phase1.snps.high_confidence.hg19.sites.vcf.idx.gz
- recalibrate SNP
#Build the SNP recalibration model
java -jar $GATK \
-T VariantRecalibrator \
-R $DB/hg19.fa \
-input raw_variants.vcf \
-resource:hapmap,known=false,training=true,truth=true,prior=15.0 hapmap.vcf \
-resource:omni,known=false,training=true,truth=true,prior=12.0 omni.vcf \
-resource:1000G,known=false,training=true,truth=false,prior=10.0 1000G.vcf \
-resource:dbsnp,known=true,training=false,truth=false,prior=2.0 dbsnp.vcf \
-an DP \ #仅适用于WGS,不适用于WES(去掉即可)
-an QD \
-an FS \
-an SOR \
-an MQ \
-an MQRankSum \
-an ReadPosRankSum \
-an InbreedingCoeff \
-mode SNP \
-tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 90.0 \
-recalFile recalibrate_SNP.recal \ #结果文件,校正后的数据,用于下一步
-tranchesFile recalibrate_SNP.tranches \ #记录命令中使用的quality score thresholds
-rscriptFile recalibrate_SNP_plots.R #提前安装好R相应的包,才能出图
#Apply the desired level of recalibration to the SNPs in the call set
java -jar $GATK \
-T ApplyRecalibration \
-R $DB/hg19.fa \
-input raw_variants.vcf \
-mode SNP \
--ts_filter_level 99.0 \ #recalibrated quality scores (VQSLOD);值越低,假阳性越低,越specific,同时丢掉真实位点的风险升高 (GATK推荐使用99.9%,provides the highest sensitivity you can get while still being acceptably specific.)这个参数依据自己需求可调,可以多次运行之后自己觉得
-recalFile recalibrate_SNP.recal \
-tranchesFile recalibrate_SNP.tranches \
-o recalibrated_snps_raw_indels.vcf #结果文件,包含所有原始数据,其中SNP多了一列recalibrated quality scores (VQSLOD),并根据是否通过过滤标注了PASS或FILTER
- recalibrate INDEL
#Build the Indel recalibration model
java -jar $GATK \
-T VariantRecalibrator \
-R $DB/hg19.fa \
-input raw_variants.vcf \
-resource:mills,known=false,training=true,truth=true,prior=12.0 Mills_and_1000G_gold_standard.indels.b37.vcf \
-resource:dbsnp,known=true,training=false,truth=false,prior=2.0 dbsnp.b37.vcf \
-an QD \
-an DP \ #仅适用于WGS,不适用于WES(去掉即可)
-an FS \
-an SOR \
-an MQRankSum \
-an ReadPosRankSum \
-an InbreedingCoeff
-mode INDEL \
-tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 90.0 \
--maxGaussians 4 \ #clusters of variants that have similar properties,WES样本数低于30时使用;样本数足够时,先去掉这个参数跑,再加上跑,然后自己选择是否要加这个参数
-recalFile recalibrate_INDEL.recal \ #结果文件,contains the recalibration data
-tranchesFile recalibrate_INDEL.tranches \
-rscriptFile recalibrate_INDEL_plots.R
#Apply the desired level of recalibration to the Indels in the call set
java -jar $GATK \
-T ApplyRecalibration \
-R $DB/hg19.fa \
-input recalibrated_snps_raw_indels.vcf \
-mode INDEL \
--ts_filter_level 99.0 \ #Best Practice推荐99.9%
-recalFile recalibrate_INDEL.recal \
-tranchesFile recalibrate_INDEL.tranches \
-o recalibrated_variants.vcf #结果文件
VariantRecalibration之前,如果缺乏相应的参数,如QD,FS,DP etc.先用VariantAnnotator获得相应的参数值。
#annotate VCF
java -jar $GATK \
-T VariantAnnotator \
-R $DB/hg19.fa \
-I input.bam \
-V raw_variants.vcf \
-o annotate_raw_variants.vcf \ #输出文件,如果遇到不符合注释的位点,就默默地略过了,可以检查header,确认做了哪些注释
--useAllAnnotations \
--dbsnp dbsnp.vcf
05 SNV初步统计分析
VQSR重在检测SNV,下一步需要使用其他信息提高genotype准确性,过滤低质量genotype,进一步精炼SNV。该流程尤其适用于以下研究:关注变异的拷贝数,家系中变异的de novo origin(https://software.broadinstitute.org/gatk/documentation/article?id=11074)。
05-1从VCF中提取子集:SelectVariants
SNP:SNPs with call rates <95% , MAF <1% or significant deviation from Hardy–Weinberg equilibrium (HWE) in controls (HWE P ≤ 1 × 10−6) were excluded.
#提取子集:SNP, biallelic, call rate>95%, filtered samples<10%
java -jar $GATK \
-T SelectVariants \
-R $DB/hg19.fa \
-ef \ #--excludeFiltered 去除被过滤的位点(在FILTER中,标注不是.或PASS)
-env \ #--excludeNonVariants 去除非变异位点
-noTrim \ #保留原始的allele
-selectType SNP \ #INDEL,SNP,MIXED等,提取某类变异
-restrictAllelesTo BIALLELIC \ #ALL,BIALLELIC,MULTIALLELIC,提取某类变异
--maxNOCALLfraction 0.05\ #Maximum fraction of samples with no-call genotypes
--maxFractionFilteredGenotypes 0.1 \ #最多有多少比例的个体被过滤掉了
-V recalibrated_variants.vcf \
-o SNP_bia.vcf
#提取子集:SNP,MULTIALLELIC, call rate>95%, filtered samples<10%
java -jar $GATK \
-T SelectVariants \
-R $DB/hg19.fa \
-ef \
-env \
-noTrim \
-selectType SNP \
-selectType MNP \
-restrictAllelesTo MULTIALLELIC \
-V recalibrated_variants.vcf \
-o SNP_multi.vcf
#提取子集:INDEL
java -jar $GATK \
-T SelectVariants \
-R $DB/hg19.fa \
-ef \
-env \
-noTrim \
-selectType INDEL \
-V recalibrated_variants.vcf \
-o indel.vcf
05-2 精炼:CalculateGenotypePosteriors VariantFiltration VariantAnnotator Funcotator (experimental)
该流程仅适用于biallelic SNP,不适用于INDEL
#Step 1: Derive posterior probabilities of genotypes
#FORMAT列会增加“PP”,同时"GQ"已更新(基于PP进行计算,方法同之前基于PL计算的GQ)
java -jar $GATK \
-T CalculateGenotypePosteriors \
-R $DB/hg19.fa \
-supporting 1000G_phase1.snps.high_confidence.hg19.sites.vcf \ #必须要有
-V SNP_bia.vcf \
-o SNP_bia.withPosteriors.vcf
#报错:Variant does not contain the same number of MLE allele counts as alternate alleles for record at chr1:94496602 说明supporting文件里在该位点是mulitallele,所以先提取出supporting文件中的biallelic位点(新文件用作supporting文件,再跑一次CalculateGenotypePosteriors)
java -jar $GATK \
-T SelectVariants \
-R $DB/hg19.fa \
-ef -env -noTrim \
-selectType SNP \
-restrictAllelesTo BIALLELIC \
-V 1000G_phase1.snps.high_confidence.hg19.sites.vcf \
-o 1000G_phase1.snps.high_confidence.hg19.sites.bia.vcf #用作supporting文件
#Step 2: Filter low quality genotypes
#genotypes with GQ < 20 based on the posteriors are filtered out.GQ20 is widely accepted as a good threshold for genotype accuracy, indicating that there is a 99% chance that the genotype in question is correct.
java -jar $GATK \
-T VariantFiltration \
-R $DB/hg19.fa \
-G_filter "GQ < 20.0" \ #用FORMAT FILED中的变量过滤
-G_filterName lowGQ \
--setFilteredGtToNocall \ #被过滤的位点,将genotype改为./.(相当于missing data
-V SNP_bia.withPosteriors.vcf \
-o SNP_bia.withPosteriors_GQ20.vcf
#Step 3: Annotate possible de novo mutations
#Tool used: VariantAnnotator 针对Family
#Step 4: Functional annotation of possible biological effects
#尚处开发阶段,类似SnpEff or Oncotator
05-3 结果评估
对结果进行评估,判断其是否符合一般规律。https://software.broadinstitute.org/gatk/documentation/article?id=11071
- Sanger sequencing
- 多个芯片结果的一致性
- 与truth set比较,评估一致性,有两个假设:truth set已经被验证过,可以看做是gold-standard; 你的样本和truth set来源的样本要有相似的遗传组成(比如都是东亚人群)
Sequencing Type # of Variants* TiTv Ratio
WGS ~4.4M 2.0-2.1
WES ~41k 3.0-3.3
*for a single sample
Variants数目:具体数值受测序类型、样本量、过滤方式等影响,如果数量级差得夸张,就说明有问题啦。
TiTv Ratio: ratio of transition (Ti) to transversion (Tv) SNPs。理论上,如果是随机突变的话,比例是0.5.但是,生物学上经常会发生甲基化,C变成T,因此Ti突变更多。而且,CpG island经常出现在primer取悦,C甲基化较多,因此WES数据更偏向Ti,比例一般在3.0-3.3。
这里就可以自己统计啦~VCF文件的处理推荐BCFtools,VCFtools,或者自己动手丰衣食足。