sra文件地址:ftp://ftp.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/
下载测序数据
# 循环下载sra文件
1,for ((i=677;i<=680;i++)) ;do wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP029/SRP029245/SRR957$i/SRR957$i.sra ;done
# 后台运行,注意括号的位置
for ((i=508;i<=523;i++)) ;do(nohup wget ftp://ftp.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR103/SRR1039$i/SRR1039$i.sra &) ;done
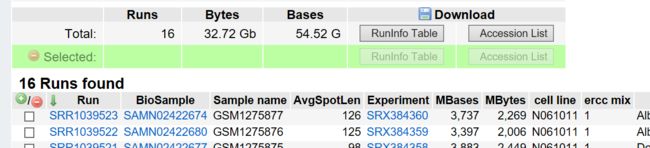
2,ncbi上获得该项目的Accession List(如图16个run代表16个数据)是一个txt文件,改名为id
image
cat id | while read id ;do prefetch $id ;done ## while read 一次读取一行
ps -ef | grep prefetch ## 查看正在运行的prefetch命令,ps -ef 相当于windows的任务管理器
ps -ef | grep prefetch | awk '{print $2}' | while read id ;do kill $id ;done ## awk提取出任务编号所在的第二列,循环杀除任务
ls | while read id ;do (nohup ../../biosoft/sratoolkit/bin/fastq-dump --gzip --split-3 -o ./ $id &) ;done ## 循环把下载的所有sra文件都变为fastq
测序数据质控
ls *.gz | while read id ;do fastqc $id ;done # 循环fastqc处理每个fastq文件
ls *.gz | xargs fastqc # 与上等效,xargs将ls的输出内容作为参数传递给fastqc,一次传递一个
multiqc ./ # 把每个数据的fastqc质控报告,合并到一个报告里,方便查看
ls *1.fastq.gz >1
ls *2.fastq.gz >2
paste 1 2 > config # 把所有的1.fastq.gz和2.fastq.gz的文件名合并到config文件
#写一个循环修剪的shell脚本,命名为qc.sh,软件为trim_galore
dir='/sas/supercloud-kong/liuhuijie/RNA-seq/clean' # 输出地址
cat $1 | while read id # $1 表示输命令时的第一个参数
do
arr=$id
fq1=${arr[0]} # config文件里一行是两个文件名,中间被空格隔开arr[0]表示第一个
fq2=${arr[1]}
nohup trim_galore -q 25 --phred33 --length 36 -e 0.1 --stringency 3 --paired -o $dir $fq1 $fq2 &
done
bash qc.sh config # 执行命令
比对,把所有的fastq数据,提取出前10000行,方便比对。
1. ls ../*.gz | while read id ;do (zcat $id | head -10000 > $(basename $id)) ;done # 提取前1万行,重新输出到$(basename $id)文件里,basename是只取文件名
2. ls ../*.gz | while read id ;do (zcat $id | head -10000 > $(basename $id '.gz')) ;done # 重输出的文件名里去除了后缀 .gz
分别用软件bowtie2、hisat2做比对得到sam文件,批量转换为bam文件
hisat2 -p 5 -x ../../index/hisat/hg38 -1 SRR1039508_1_val_1.fq -2 SRR1039508_2_val_2.fq -S tmp.hisat.sam
bowtie2 -p 5 -x ../../index/bowie/hg38 -1 SRR1039508_1_val_1.fq -2 SRR1039508_2_val_2.fq > tmp.bowtie.sam
ls *.sam | while read id ;do (samtools sort -O bam -@ 5 -o $(basename $id '.sam').bam $id) ;done # 批量转换为bam,把后缀.sam去掉换成bam
samtools view tmp.bowtie.bam | less -S #查看bam文件,可试试IGV可视化查看
ls *.bam | xargs -i samtools index {} #批量为bam文件建索引,xargs -i把bam文件作为参数传递给samtools,{}代替了bam文件
ls *bam | while read id ;do (samtools flagstat -@ 10 $id > $(basename $id '.bam').flagstat) ;done # flagstat 统计
cat *flagstat | cut -d ' ' -f 1 | paste - - - - - - - - - - - - - # 合并所有的flagstat文件的第一列,每个flagstat文件都为13行,有13个-
把当前文件夹里的fastq数据,用hisat2和bowtie软件,按顺序比对到hg38
ls *gz | cut -d "_" -f 1 | sort -u | while read id ;do (ls -lh ${id}_1_val_1.fq.gz ${id}_2_val_2.fq.gz | hisat2 -p 5 -x ../index/hisat/hg38 -1 ${id}_1_val_1.fq.gz -2 ${id}_2_val_2.fq.gz -S $id.hisat.sam);done
ls *gz | cut -d "_" -f 1 | sort -u | while read id ;do (ls -lh ${id}_1_val_1.fq.gz ${id}_2_val_2.fq.gz | bowtie2 -p 5 -x ../index/bowtie/hg38 -1 ${id}_1_val_1.fq.gz -2 ${id}_2_val_2.fq.gz > $id.bowtie.sam);done
定量
nohup featureCounts -T 10 -p -t exon -g gene_id -a ../gtf/Homo_sapiens.GRCh38.92.chr.gtf.gz -o all.id.txt *bam 1>count.log 2>&1 &
# featureCounts 进行定量,统计比对在这个基因的坐标上的read数
multiqc all.id.txt.summary #用multiqx对上步得到的结果进行可视化
表达矩阵探索
rm(list = ls())
options(stringsAsFactors = F)
a= read.table('all.id.txt',header = T) #header
meta=a[,1:6]
exprSet=a[,7:ncol(a)]
a2=exprSet[,'SRR1039508.hisat.bam'] ##
library(pheatmap)
png('heatmap.png')
corrplot(cor(exprSet))
pheatmap(scale(cor(log2(exprSet+1))))
dev.off()
##
library(airway)
data("airway")
exprSet=assay(airway)
a1=exprSet[,'SRR1039508'] ##
group_list=colData(airway)[,3]
##hclust, 层次聚类包
colnames(exprSet)=paste(group_list,1:ncol(exprSet),sep = '_')
##difine nodePar
nodePar=list(lab.cex=0.6, pch=c(NA, 19),cex=0.7,col='blue')
hc=hclust(dist(t(log2(exprSet+1))))
par(mar=c(5,5,5,10)) #par函数设置图形边距,mar参数设置边距
png('hclust.png',res = 120)
plot(as.dendrogram(hc),nodePar=nodePar,horiz=TRUE)
dev.off()
a2=data.frame(id=meta[,1],a2=a2)
a1=data.frame(id=names(a1),a1)
library(stringr) #stringr包处理字符串
a2$id=str_split(a2$id,'\\.',simplify = T)[,1]
tmp=merge(a1,a2,by='id')
png('tmp.png')
plot(tmp[,c(2,4)]) #从图上看更直观
dev.off()
DEseq2筛选差异表达基因
library(DESeq2)
library(edgeR)
library(limma)
library(airway)
data("airway")
exprSet=assay(airway) #定量后的信息
group_list=colData(airway)[,3] #提取出来了分组信息,也可以手动写成c()
colData=data.frame(row.names = colnames(exprSet),
group=group_list)
dds=DESeqDataSetFromMatrix(countData = exprSet,
colData = colData,
design = ~group) #获取矩阵信息
dds=DESeq(dds)
res=results(dds,contrast = c('group','trt','untrt'))
summary(res) #利用summary命令统计显示一共多少个genes上调和下调
resOrdered=res[order(res$padj),] #根据padj(p值经过多重校验校正后的值)排序
DEG=as.data.frame(resOrdered)
DEG=na.omit(DEG)
library(pheatmap)
choose_gene=head(rownames(DEG),100)
choose_matrix=exprSet[choose_gene,] #抽取差异表达显著的前100个基因
choose_matrix=t(scale(t(choose_matrix))) #用t函数转置,scale函数标准化
pheatmap(choose_matrix,filename='DEG_top100.png')
火山图
logFC_cutoff=with(DEG,mean(abs(log2FoldChange))+2*sd(abs(log2FoldChange))) #算log2FoldChange的阈值,with 提取数据框中的某些参数做运算,abs求绝对值,sd求标准差
DEG$change=as.factor(ifelse(DEG$pvalue<0.05 & abs(DEG$log2FoldChange)>logFC_cutoff,ifelse(DEG$log2FoldChange>logFC_cutoff,'UP','DOWN'),'NOT'))
#ifelse函数,大于logFC_cutoff的设为up,小于为down
this_title=paste0('Cutoff for logFC is ',round(logFC_cutoff,3), '\nThe number of up gene is ',nrow(DEG[DEG$change=='UP',]), '\nThe number of down gene is ',nrow(DEG[DEG$change=='DOWN',])) #paste0函数,默认是sep=""
library(ggplot2)
g=ggplot(data = DEG,
aes(x=log2FoldChange,y=-log10(pvalue),
color=change))+
geom_point(alpha=0.4,size=1.75)+
theme_set(theme_set(theme_bw(base_size = 20)))+
xlab('log2 fold change')+ylab('-log10 p-value')+
ggtitle(this_title)+theme(plot.title = element_text(size = 15,hjust = 0.5))+
scale_color_manual(values = c('blue','black','red')) #corresponding to the levels(res$change)
ggsave(g,filename = 'volcano.png')
png('dispersions.png',1000,1000,pointsize = 20)
plotDispEsts(dds,main='dispersion plot')
dev.off()
作图查看原始定量后的数据和normalization后的数据的差异
rld=rlogTransformation(dds) #DEseq2自带的rlog算法对数据进行count矩阵转换
exprMatrix_rlog=assay(rld)
png('DEseq_RAWvsNORM.png',height = 800,width = 800)
par(cex=0.7) #par函数设定全局绘图参数,cex放大多少倍
n.sample=ncol(exprSet)
if(n.sample>40) par(cex=0.5) #
cols=rainbow(n.sample*1.2) #rainbow 渐变的彩虹色
par(mfrow=c(2,2)) #4个图按行排序
boxplot(exprSet, col=cols,main='expression value',las=2) #las为2,标签垂直坐标轴
boxplot(exprMatrix_rlog, col=cols, main='expression value',las=2)
hist(as.matrix(exprSet))
hist(exprMatrix_rlog)
dev.off()