- python 自动化数据提取之正则表达式_python 正则提取(2)
m0_60607245
程序员python学习面试
一、Python所有方向的学习路线Python所有方向的技术点做的整理,形成各个领域的知识点汇总,它的用处就在于,你可以按照下面的知识点去找对应的学习资源,保证自己学得较为全面。二、Python必备开发工具工具都帮大家整理好了,安装就可直接上手!三、最新Python学习笔记当我学到一定基础,有自己的理解能力的时候,会去阅读一些前辈整理的书籍或者手写的笔记资料,这些笔记详细记载了他们对一些技术点的理
- 如何解决分布式应用数量庞大而导致数据库连接数满的问题?
纵然间
数据库
修改数据库服务器的配置文件或参数来增加最大连接数限制。例如,在MySQL中,可以通过修改my.cnf(Linux)或my.ini(Windows)文件中的max_connections参数来增加最大连接数。具体的操作方法可以参考数据库服务器的官方文档或相关技术支持。检查应用程序代码,确保在使用完数据库连接后及时释放连接资源,避免长时间占用连接而导致连接数不足。可以使用连接池技术来管理数据库连接,提
- Mysql学习笔记-Mysql基础进阶
少年无为
MysqlMysql数据库多表查询数据库备份Mysql查询
#知识点1.DQL:查询语句1.排序查询2.聚合函数3.分组查询4.分页查询2.约束3.多表之间的关系4.范式5.数据库的备份和还原#DQL:查询语句1.排序查询*语法:orderby子句*orderby排序字段1排序方式1,排序字段2排序方式2...*排序方式:*ASC:升序,默认的。*DESC:降序。*注意:*如果有多个排序条件,则当前边的条件值一样时,才会判断第二条件。2.聚合函数:将一列数
- ADC(模数转换器)与DAC(数模转换器)详解:从基础到应用示例
楼台的春风
嵌入式开发STM32嵌入式c语言mcu自动驾驶嵌入式硬件stm32物联网
ADC(模数转换器)与DAC(数模转换器)详解:从基础到应用示例目录ADC(模数转换器)与DAC(数模转换器)详解:从基础到应用示例引言一、ADC(模数转换器)1.ADC的基本概念2.ADC的工作原理3.ADC的主要类型4.ADC的技术指标5.ADC的应用场景6.ADC在嵌入式系统中的使用案例二、DAC(数模转换器)1.DAC的基本概念2.DAC的工作原理3.DAC的主要类型4.DAC的技术指标5
- 内外网隔离文件传输解决方案|系统与钉钉集成+等保合规,安全提升70%
CSTechAI
钉钉安全中间件安全架构
内外网隔离文件传输解决方案|系统与钉钉集成+等保合规,安全提升70%##一、背景与痛点在内外网隔离的企业网络环境中,员工与外部协作伙伴(如钉钉用户)的文件传输面临以下挑战:1.**安全性风险**:内外网直连可能导致病毒传播、数据泄露。2.**操作繁琐**:传统方式需频繁切换网络环境,降低工作效率。3.**审计缺失**:缺乏文件传输的完整日志记录,难以追溯责任。**系统**通过智能中转架构,在保障网
- Jmeter 性能-稳定性测试TPS计算
软件测试媛
软件测试技术分享自动化测试jmeter软件测试功能测试
1、普通计算公式TPS=总请求数/总时间1按照需求得到基础数据,比如在去年第xxx周,某平台有5万的浏览量那么总请求数我们可以估算为5万(1次浏览都至少对应1个请求)总请求数=50000请求数总时间:由于不知道每个请求的具体时间,按照普通方法,可以按照一天的时间进行计算总时间=1天=1*24小时=24*36001秒套入公式可得:TPS=50000/24*3600秒=0.58tps1结论:按照普通计
- Python从0到100(三十九):数据提取之正则(文末免费送书)
是Dream呀
pythonmysql开发语言
前言:零基础学Python:Python从0到100最新最全教程。想做这件事情很久了,这次我更新了自己所写过的所有博客,汇集成了Python从0到100,共一百节课,帮助大家一个月时间里从零基础到学习Python基础语法、Python爬虫、Web开发、计算机视觉、机器学习、神经网络以及人工智能相关知识,成为学习学习和学业的先行者!欢迎大家订阅专栏:零基础学Python:Python从0到100最新
- ollama的docker 使用教程
贾斯汀玛尔斯
数据湖AIDocker容器dockereureka容器
好的,下面是Ollama在Docker中的使用教程。我将详细描述如何在Docker容器中运行Ollama,包括安装、配置和常用操作。OllamaDocker使用教程Ollama可以通过Docker运行,提供了一个简洁且隔离的环境来使用AI模型。本文将引导你如何在Docker中设置和使用Ollama。目录前提条件拉取OllamaDocker镜像启动Ollama容器基本命令操作停止容器<
- docker部署kafka(单节点) + Springboot集成kafka
wsdhla
dockerkafkaspringbootzookeeper
环境:操作系统:win10Docker:DockerDesktop4.21.1(114176)、DockerEnginev24.0.2SpringBoot:2.7.15步骤1:创建网络:dockernetworkcreate--subnet=172.18.0.0/16net-kafka步骤2:安装zk镜像dockerpullzookeeper:latestdockerrun-d--restarta
- mysql实时同步到es
数据库
测试了多个方案同步,最终选择oceanu产品,底层基于Flinkcdc1、实时性能够保证,binlog量很大时也不产生延迟2、配置SQL即可完成,操作上简单下面示例mysql的100张分表实时同步到es,优化备注等文本字段的like查询创建SQL作业CREATETABLEfrom_mysql(idint,cidintNOTNULL,gidbigintNOTNULL,contentvarchar,c
- YashanDB数据分区
数据库
本文内容来自YashanDB官网,原文内容请见https://doc.yashandb.com/yashandb/23.3/zh/%E6%A6%82%E5%BF%B5%...#分区概述YashanDB可以将大规模数据拆分成更小、更便于管理的对象,即分区。通过对数据进行分区管理,可以减少无效数据的访问,提升大规模数据下的访问、操作性能。表可以根据某些条件进行分区,不同分区独立管理。分区表提供了更高效
- 深度学习环境配置——Anaconda安装
tyyhmtyyhm
深度学习环境配置深度学习人工智能
目录Ⅰ.Windows系统安装Anaconda1.1下载安装Ⅱ.Linux系统安装Anaconda(适用于服务器安装)2.1下载2.2安装操作系统:windows11/ubuntu20/ubuntu18更新时间:20240221Ⅰ.Windows系统安装Anaconda1.1下载安装https://www.anaconda.com/download默认安装即可。Ⅱ.Linux系统安装Anacond
- 使用Arcgis API for android加载OpenStreetMap底图并完成定位
续汉冕
移动开发androidandroidstudioarcgisapi
为了完成这个应用功能花了三天,代码倒不多就是比较坑!环境:AndroidSDKAPI22,AndroidStudio1.2.2,arcgisandroidSDK10.2.7如何基于ArcgisAPIforandroid在AndroidStudio新建一个项目就不再赘述了,大家可以参考以下网址:使用AndroidStudio与ArcgisandroidSDK的开发环境部署和HelloWorld:ht
- 深度学习工厂的蓝图:拆解CUDA驱动、PyTorch与OpenCV的依赖关系
时光旅人01号
深度学习pytorchopencv
想象一下,你正在建造一座深度学习工厂,这座工厂专门用于高效处理深度学习任务(如训练神经网络)和计算机视觉任务(如图像处理)。为了让工厂顺利运转,你需要搭建基础设施、安装设备、设置生产线,并配备控制台来管理整个生产过程。以下是这座工厂的详细构建过程:1.工厂的基础设施:Ubuntu比喻:Ubuntu是工厂所在的土地和建筑,提供了基础设施和运行环境。作用:提供操作系统环境,支持安装和运行各种工具和框架
- FakeApp 技术浅析(一)
爱研究的小牛
AIGC—深度伪造虚拟现实人工智能AIGC深度学习机器学习
FakeApp是一款早期的深度伪造(Deepfake)工具,最初于2018年发布,用于生成和编辑换脸视频。尽管FakeApp已经不再更新,但它在深度伪造技术的发展中起到了重要作用。1.技术背景与理论基础1.1生成对抗网络(GANs)生成对抗网络(GANs)是深度学习领域中的一种重要模型,由生成器(Generator)和判别器(Discriminator)组成。生成器负责生成逼真的数据(如图像、视频
- USB转串口芯片CH9102替代CP2102注意事项
Chery1140
单片机嵌入式硬件
CH9102与CP2102可实现pin2pin兼容,可以在不更改硬件设计的前提下实现不同型号间快速切换与产品应用。CH9102系列型号包括:CH9102F(QFN24)和CH9102X(QFN28),CP2102系列型号包括:CP2102、CP2102N-GQFN24、CP2102N-GQFN28。1.应用差异说明1)驱动说明:CH9102芯片为CDC类串口芯片,用户可以选择使用操作系统内置的CD
- 网络协议、网络安全架构、网络安全标准
Utopia.️
网络协议web安全架构
1.网络协议网络协议是计算机网络中设备之间通信的规则集。熟悉常见的网络协议及其工作原理是确保网络安全的基础。常见协议:TCP/IP协议:这是网络通信的基础协议,确保数据从源端传输到目标端,支持多种传输方式(TCP可靠传输,UDP快速但不可靠)。HTTP/HTTPS:HTTP用于浏览器与服务器之间的通信,HTTPS则是在HTTP上添加了SSL/TLS加密层,用于确保数据传输的安全性。DNS协议:用于
- 嵌入式音视频开发(二)ffmpeg音视频同步
云雨歇
音视频ffmpeg
系列文章目录嵌入式音视频开发(零)移植ffmpeg及推流测试嵌入式音视频开发(一)ffmpeg框架及内核解析嵌入式音视频开发(二)ffmpeg音视频同步嵌入式音视频开发(三)直播协议及编码器文章目录系列文章目录前言一、音视频同步1.1基础概念1.2三种同步方法二、音视频同步的实现2.1时间基的转换问题2.2音频为基准2.2.1实现思路2.2.2代码大纲2.3外部时钟同步2.3.1实现思路2.3.2
- JMM(Java内存模型)讲解
十五001
基础javajvm
JMM(JavaMemoryModel,Java内存模型)是Java并发编程中的一个非常重要的概念,它帮助我们理解Java程序在多线程环境下内存操作的行为。别担心,我会用简单易懂的方式来讲解,让你轻松掌握它的核心内容。1.什么是JMM?定义JMM是Java内存模型的简称,它定义了Java程序中内存操作的规则和规范。简单来说,JMM规定了Java程序中的变量存储在内存中的方式,以及线程如何读取和写入
- 麒麟v10安装mysql5.7(ARM架构)
qqxinxi
arm开发
下载路径:华为云镜像麒麟v10是潮流时代的新时髦的linux操作系统,但随着ARM架构流行,出现了一些卡点,不以为然,没当回事的大吃一惊。经常卡住。例如:在安装mysql5.7(ARM架构)最简单:使用rpmmysql-5.7.27.1.el7.aarch64.rpm文件比较小下载完之后rpm-ivhmysql-5.7.27.1.el7.aarch64.rpm比较简单常用的方法,再不能连接互联网时
- TK群发器:提升TikTok营销效率的智能工具
@ V:ZwaitY09
矩阵tiktok
随着短视频平台TikTok的快速发展,许多企业和内容创作者都将其作为重要的营销渠道。但随着平台的竞争加剧,如何高效管理多个账号、提升曝光度和互动率,成为了营销者的一大挑战。为了解决这一问题,TK群发器应运而生。它通过智能化的操作方式,帮助用户精准高效地进行多账号管理和内容群发,极大提高了营销效率。TK群发器的主要功能:多账号精准群发:TK群发器支持同时管理多个TikTok账号,用户可以通过该工具实
- OpenLayers总结3
Super毛毛穗
WebGIS开发OpenLayersGISWebGIS
一、静态测距1.原理静态测距主要是针对地图上已有的矢量要素(如线要素),利用OpenLayers提供的几何计算函数来获取其长度。在实际操作中,先加载包含几何要素的GeoJSON数据到矢量图层,当鼠标指针移动到要素上时,获取该要素的几何信息,再调用getLength函数计算其长度。2.代码实现步骤及注释//引入必要的模块importVectorLayerfrom"ol/layer/Vector.js
- 【CUDA】Pytorch_Extensions
joker D888
深度学习pytorchpythoncudac++深度学习
【CUDA】Pytorch_Extensions为什么要开发CUDA扩展?当我们在PyTorch中实现自定义算子时,通常有两种选择:使用纯Python实现(简单但效率低)使用C++/CUDA扩展(高效但需要编译)对于计算密集型的操作(如神经网络中的自定义激活函数),使用CUDA扩展可以获得接近硬件极限的性能。本文将以实现一个多项式激活函数x²+x+1为例,展示完整的开发流程。完整CUDA扩展代码解
- centos操作系统安装R包单细胞拟时序分析CytoTRACE2
探序基因
centoslinux运维
探序基因肿瘤研究院整理作者操作系统为centosstream8,R版本为4.3.3devtools::install_github("digitalcytometry/cytotrace2",subdir="cytotrace2_r")中途出现错误:*installing*source*package‘RcppGSL’...**成功将‘RcppGSL’程序包解包并MD5和检查**usingstag
- 基于python使用scanpy分析单细胞转录组数据
探序基因
单细胞分析python开发语言
探序基因肿瘤研究院整理相关后缀的格式介绍:.h5ad:是一种用于存储单细胞数据的文件格式,可以通过anndata库在Python中处理.loom:高效的数据存储格式(.loom文件),使得用户可以轻松地存储、查询和分析大规模的单细胞数据集。Loompy的设计目标是提供一个快速、灵活且易于使用的工具,以支持生物信息学家和研究人员在单细胞水平上进行数据分析。python的单细胞转录组数据结构说明:da
- GATK3.5GATK4.0与java版本的关系
探序基因
java
探序基因肿瘤研究院整理操作系统centosstream9yum安装java后,输入java-version可看到:openjdkversion"11.0.20.1"2023-08-24LTSOpenJDKRuntimeEnvironment(Red_Hat-11.0.20.1.1-2)(build11.0.20.1+1-LTS)OpenJDK64-BitServerVM(Red_Hat-11.0.
- 位图(BitMap)实现
小猫猫猫◍˃ᵕ˂◍
bitmap算法
位图(BitMap)实现1.位图简介位图(BitMap)是一种高效的数据结构,用于存储和操作位(bit)数据。每个位可以表示一个布尔值(0或1),常用于去重、排序、快速查找等场景。2.核心功能⚙️设置位(Set):将某一位设置为1。清除位(Clear):将某一位设置为0。获取位(Get):检查某一位是否为1。打印位图(Print):以二进制形式打印位图。3.代码实现packageMyStruct;
- Mybatis判断问题:深入解析与实战案例
DTcode7
sql数据库相关数据库mysqlSQL数据库开发sql
Mybatis判断问题:深入解析与实战案例基础概念与作用说明``标签``,``,````示例一:基本的``标签使用说明示例二:``,``,``的使用说明示例三:使用``标签简化条件语句说明实际工作中的使用技巧自行拓展内容在现代企业级应用开发中,MyBatis作为一款优秀的持久层框架,以其灵活的SQL映射机制和强大的动态SQL功能,深受广大开发者的喜爱。然而,在使用过程中,如何准确地进行条件判断,特
- 在线预览 Word 文档
你不讲 wood
word开发语言前端vue.jsjavascriptnode.jsdocx-preview
引言随着互联网技术的发展,Web应用越来越复杂,用户对在线办公的需求也日益增加。在许多业务场景中,能够直接在浏览器中预览Word文档是一个非常实用的功能。这不仅可以提高用户体验,还能减少用户操作步骤,提升效率。实现原理1.后端服务假设后端服务已经提供了两个API接口:getFilesList:获取文件列表。previewFile:获取指定文件的内容。constexpress=require('ex
- 前端导出word文件—包含canvas(echarts图表)
Liuer_Qin
jscanvasechartsecharts前端javascript
一、使用的插件html-docx-js二、整体思路因为canvas是运行在内存中的,所以不能简单的通过dom获取canvas图片,需要手动的先将canvas转为image。三、实现先克隆要下载的DOM的副本。因为canvas是运行在内存中的,所以也不能通过cloneNode方法克隆下来(克隆下来是空的)。我们这里将原DOM中的canvas转成图片,然后插入到副本的对应位置,这样操作不会影响原DOM
- log4j对象改变日志级别
3213213333332132
javalog4jlevellog4j对象名称日志级别
log4j对象改变日志级别可批量的改变所有级别,或是根据条件改变日志级别。
log4j配置文件:
log4j.rootLogger=ERROR,FILE,CONSOLE,EXECPTION
#log4j.appender.FILE=org.apache.log4j.RollingFileAppender
log4j.appender.FILE=org.apache.l
- elk+redis 搭建nginx日志分析平台
ronin47
elasticsearchkibanalogstash
elk+redis 搭建nginx日志分析平台
logstash,elasticsearch,kibana 怎么进行nginx的日志分析呢?首先,架构方面,nginx是有日志文件的,它的每个请求的状态等都有日志文件进行记录。其次,需要有个队 列,redis的l
- Yii2设置时区
dcj3sjt126com
PHPtimezoneyii2
时区这东西,在开发的时候,你说重要吧,也还好,毕竟没它也能正常运行,你说不重要吧,那就纠结了。特别是linux系统,都TMD差上几小时,你能不痛苦吗?win还好一点。有一些常规方法,是大家目前都在采用的1、php.ini中的设置,这个就不谈了,2、程序中公用文件里设置,date_default_timezone_set一下时区3、或者。。。自己写时间处理函数,在遇到时间的时候,用这个函数处理(比较
- js实现前台动态添加文本框,后台获取文本框内容
171815164
文本框
<%@ page language="java" import="java.util.*" pageEncoding="UTF-8"%>
<!DOCTYPE html PUBLIC "-//W3C//DTD XHTML 1.0 Transitional//EN" "http://w
- 持续集成工具
g21121
持续集成
持续集成是什么?我们为什么需要持续集成?持续集成带来的好处是什么?什么样的项目需要持续集成?... 持续集成(Continuous integration ,简称CI),所谓集成可以理解为将互相依赖的工程或模块合并成一个能单独运行
- 数据结构哈希表(hash)总结
永夜-极光
数据结构
1.什么是hash
来源于百度百科:
Hash,一般翻译做“散列”,也有直接音译为“哈希”的,就是把任意长度的输入,通过散列算法,变换成固定长度的输出,该输出就是散列值。这种转换是一种压缩映射,也就是,散列值的空间通常远小于输入的空间,不同的输入可能会散列成相同的输出,所以不可能从散列值来唯一的确定输入值。简单的说就是一种将任意长度的消息压缩到某一固定长度的消息摘要的函数。
- 乱七八糟
程序员是怎么炼成的
eclipse中的jvm字节码查看插件地址:
http://andrei.gmxhome.de/eclipse/
安装该地址的outline 插件 后重启,打开window下的view下的bytecode视图
http://andrei.gmxhome.de/eclipse/
jvm博客:
http://yunshen0909.iteye.com/blog/2
- 职场人伤害了“上司” 怎样弥补
aijuans
职场
由于工作中的失误,或者平时不注意自己的言行“伤害”、“得罪”了自己的上司,怎么办呢?
在职业生涯中这种问题尽量不要发生。下面提供了一些解决问题的建议:
一、利用一些轻松的场合表示对他的尊重
即使是开明的上司也很注重自己的权威,都希望得到下属的尊重,所以当你与上司冲突后,最好让不愉快成为过去,你不妨在一些轻松的场合,比如会餐、联谊活动等,向上司问个好,敬下酒,表示你对对方的尊重,
- 深入浅出url编码
antonyup_2006
应用服务器浏览器servletweblogicIE
出处:http://blog.csdn.net/yzhz 杨争
http://blog.csdn.net/yzhz/archive/2007/07/03/1676796.aspx
一、问题:
编码问题是JAVA初学者在web开发过程中经常会遇到问题,网上也有大量相关的
- 建表后创建表的约束关系和增加表的字段
百合不是茶
标的约束关系增加表的字段
下面所有的操作都是在表建立后操作的,主要目的就是熟悉sql的约束,约束语句的万能公式
1,增加字段(student表中增加 姓名字段)
alter table 增加字段的表名 add 增加的字段名 增加字段的数据类型
alter table student add name varchar2(10);
&nb
- Uploadify 3.2 参数属性、事件、方法函数详解
bijian1013
JavaScriptuploadify
一.属性
属性名称
默认值
说明
auto
true
设置为true当选择文件后就直接上传了,为false需要点击上传按钮才上传。
buttonClass
”
按钮样式
buttonCursor
‘hand’
鼠标指针悬停在按钮上的样子
buttonImage
null
浏览按钮的图片的路
- 精通Oracle10编程SQL(16)使用LOB对象
bijian1013
oracle数据库plsql
/*
*使用LOB对象
*/
--LOB(Large Object)是专门用于处理大对象的一种数据类型,其所存放的数据长度可以达到4G字节
--CLOB/NCLOB用于存储大批量字符数据,BLOB用于存储大批量二进制数据,而BFILE则存储着指向OS文件的指针
/*
*综合实例
*/
--建立表空间
--#指定区尺寸为128k,如不指定,区尺寸默认为64k
CR
- 【Resin一】Resin服务器部署web应用
bit1129
resin
工作中,在Resin服务器上部署web应用,通常有如下三种方式:
配置多个web-app
配置多个http id
为每个应用配置一个propeties、xml以及sh脚本文件
配置多个web-app
在resin.xml中,可以为一个host配置多个web-app
<cluster id="app&q
- red5简介及基础知识
白糖_
基础
简介
Red5的主要功能和Macromedia公司的FMS类似,提供基于Flash的流媒体服务的一款基于Java的开源流媒体服务器。它由Java语言编写,使用RTMP作为流媒体传输协议,这与FMS完全兼容。它具有流化FLV、MP3文件,实时录制客户端流为FLV文件,共享对象,实时视频播放、Remoting等功能。用Red5替换FMS后,客户端不用更改可正
- angular.fromJson
boyitech
AngularJSAngularJS 官方APIAngularJS API
angular.fromJson 描述: 把Json字符串转为对象 使用方法: angular.fromJson(json); 参数详解: Param Type Details json
string
JSON 字符串 返回值: 对象, 数组, 字符串 或者是一个数字 示例:
<!DOCTYPE HTML>
<h
- java-颠倒一个句子中的词的顺序。比如: I am a student颠倒后变成:student a am I
bylijinnan
java
public class ReverseWords {
/**
* 题目:颠倒一个句子中的词的顺序。比如: I am a student颠倒后变成:student a am I.词以空格分隔。
* 要求:
* 1.实现速度最快,移动最少
* 2.不能使用String的方法如split,indexOf等等。
* 解答:两次翻转。
*/
publ
- web实时通讯
Chen.H
Web浏览器socket脚本
关于web实时通讯,做一些监控软件。
由web服务器组件从消息服务器订阅实时数据,并建立消息服务器到所述web服务器之间的连接,web浏览器利用从所述web服务器下载到web页面的客户端代理与web服务器组件之间的socket连接,建立web浏览器与web服务器之间的持久连接;利用所述客户端代理与web浏览器页面之间的信息交互实现页面本地更新,建立一条从消息服务器到web浏览器页面之间的消息通路
- [基因与生物]远古生物的基因可以嫁接到现代生物基因组中吗?
comsci
生物
大家仅仅把我说的事情当作一个IT行业的笑话来听吧..没有其它更多的意思
如果我们把大自然看成是一位伟大的程序员,专门为地球上的生态系统编制基因代码,并创造出各种不同的生物来,那么6500万年前的程序员开发的代码,是否兼容现代派的程序员的代码和架构呢?
- oracle 外部表
daizj
oracle外部表external tables
oracle外部表是只允许只读访问,不能进行DML操作,不能创建索引,可以对外部表进行的查询,连接,排序,创建视图和创建同义词操作。
you can select, join, or sort external table data. You can also create views and synonyms for external tables. Ho
- aop相关的概念及配置
daysinsun
AOP
切面(Aspect):
通常在目标方法执行前后需要执行的方法(如事务、日志、权限),这些方法我们封装到一个类里面,这个类就叫切面。
连接点(joinpoint)
spring里面的连接点指需要切入的方法,通常这个joinpoint可以作为一个参数传入到切面的方法里面(非常有用的一个东西)。
通知(Advice)
通知就是切面里面方法的具体实现,分为前置、后置、最终、异常环
- 初一上学期难记忆单词背诵第二课
dcj3sjt126com
englishword
middle 中间的,中级的
well 喔,那么;好吧
phone 电话,电话机
policeman 警察
ask 问
take 拿到;带到
address 地址
glad 高兴的,乐意的
why 为什么
China 中国
family 家庭
grandmother (外)祖母
grandfather (外)祖父
wife 妻子
husband 丈夫
da
- Linux日志分析常用命令
dcj3sjt126com
linuxlog
1.查看文件内容
cat
-n 显示行号 2.分页显示
more
Enter 显示下一行
空格 显示下一页
F 显示下一屏
B 显示上一屏
less
/get 查询"get"字符串并高亮显示 3.显示文件尾
tail
-f 不退出持续显示
-n 显示文件最后n行 4.显示头文件
head
-n 显示文件开始n行 5.内容排序
sort
-n 按照
- JSONP 原理分析
fantasy2005
JavaScriptjsonpjsonp 跨域
转自 http://www.nowamagic.net/librarys/veda/detail/224
JavaScript是一种在Web开发中经常使用的前端动态脚本技术。在JavaScript中,有一个很重要的安全性限制,被称为“Same-Origin Policy”(同源策略)。这一策略对于JavaScript代码能够访问的页面内容做了很重要的限制,即JavaScript只能访问与包含它的
- 使用connect by进行级联查询
234390216
oracle查询父子Connect by级联
使用connect by进行级联查询
connect by可以用于级联查询,常用于对具有树状结构的记录查询某一节点的所有子孙节点或所有祖辈节点。
来看一个示例,现假设我们拥有一个菜单表t_menu,其中只有三个字段:
- 一个不错的能将HTML表格导出为excel,pdf等的jquery插件
jackyrong
jquery插件
发现一个老外写的不错的jquery插件,可以实现将HTML
表格导出为excel,pdf等格式,
地址在:
https://github.com/kayalshri/
下面看个例子,实现导出表格到excel,pdf
<html>
<head>
<title>Export html table to excel an
- UI设计中我们为什么需要设计动效
lampcy
UIUI设计
关于Unity3D中的Shader的知识
首先先解释下Unity3D的Shader,Unity里面的Shaders是使用一种叫ShaderLab的语言编写的,它同微软的FX文件或者NVIDIA的CgFX有些类似。传统意义上的vertex shader和pixel shader还是使用标准的Cg/HLSL 编程语言编写的。因此Unity文档里面的Shader,都是指用ShaderLab编写的代码,
- 如何禁止页面缓存
nannan408
htmljspcache
禁止页面使用缓存~
------------------------------------------------
jsp:页面no cache:
response.setHeader("Pragma","No-cache");
response.setHeader("Cache-Control","no-cach
- 以代码的方式管理quartz定时任务的暂停、重启、删除、添加等
Everyday都不同
定时任务管理spring-quartz
【前言】在项目的管理功能中,对定时任务的管理有时会很常见。因为我们不能指望只在配置文件中配置好定时任务就行了,因为如果要控制定时任务的 “暂停” 呢?暂停之后又要在某个时间点 “重启” 该定时任务呢?或者说直接 “删除” 该定时任务呢?要改变某定时任务的触发时间呢? “添加” 一个定时任务对于系统的使用者而言,是不太现实的,因为一个定时任务的处理逻辑他是不
- EXT实例
tntxia
ext
(1) 增加一个按钮
JSP:
<%@ page language="java" import="java.util.*" pageEncoding="UTF-8"%>
<%
String path = request.getContextPath();
Stri
- 数学学习在计算机研究领域的作用和重要性
xjnine
Math
最近一直有师弟师妹和朋友问我数学和研究的关系,研一要去学什么数学课。毕竟在清华,衡量一个研究生最重要的指标之一就是paper,而没有数学,是肯定上不了世界顶级的期刊和会议的,这在计算机学界尤其重要!你会发现,不论哪个领域有价值的东西,都一定离不开数学!在这样一个信息时代,当google已经让世界没有秘密的时候,一种卓越的数学思维,绝对可以成为你的核心竞争力. 无奈本人实在见地


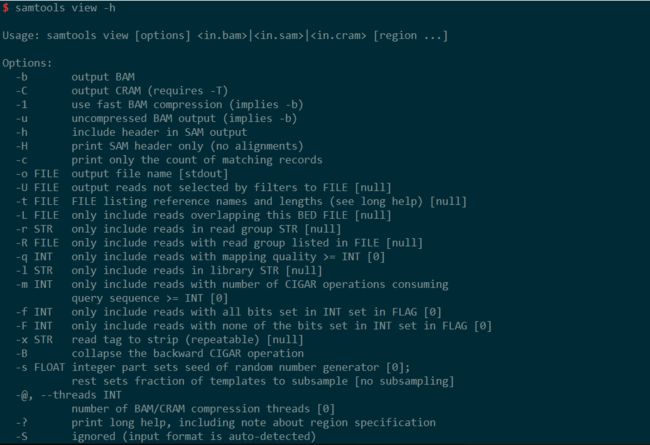
 less看sam的head
less看sam的head