「论文翻译」Graph convolutional networks for computational drug development and discovery
briefings in Bioinformatics 2019 (B类)
文章目录
- Abstract
- Introduction
- Principles of graph convolution
- Application of GCN on drug discovery
- Quantitative Structure Activity/Property Relationship Prediction
- Biological property and activity
- Quantum mechanical property
- Incorporate GCN with other learning architecture
- Interaction prediction
- Ligand–protein (drug–target) interaction
- Protein–protein interaction
- Drug–drug interaction
- Synthesis prediction
- De novo molecular design
- Databases for drug discovery and molecular bioinformatics
- Molecular property and activity
- Biological property and activity
- Quantum chemical property
- Interaction database
- Synthesis database
- Integrated benchmark database
- Discussion
- Database challenges and opportunities
- Methodology challenges and opportunities
- Network design challenges and opportunities
- Interpretability challenges and opportunities
Abstract
尽管在过去十年中深度学习在各个领域都取得了令人瞩目的成功,但其在分子信息学和药物发现中的应用仍然受到限制。在深层架构适应结构化数据方面的最新进展为药物研究开辟了新的范例。在这项调查中,我们对图卷积网络的领域以及图卷积网络在药物发现和分子信息学中的应用进行了系统的综述。通常,我们对图卷积网络为何以及如何可以帮助完成与药物有关的任务感兴趣。我们通过四个角度阐述了现有的应用:分子性质和活性预测,相互作用预测,合成预测和de novo药物设计。我们简要介绍图卷积网络背后的理论基础,并说明基于不同公式的各种体系结构。然后,我们总结了药物相关问题中的代表性应用。我们还将讨论将图卷积网络应用于药物发现的当前挑战和未来可能性。
Introduction
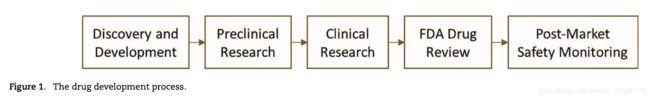
药物开发是一个昂贵且耗时的过程,其中要测试成千上万种化合物并进行实验以找出安全有效的药物。 药物开发的一般过程涉及如Fig. 1所示的步骤。现代药物开发旨在加快中间步骤,从而通过在药物开发和临床前研究阶段利用机器学习工具进行药物开发来降低成本。简而言之,通过逐步进行的一系列测试对分子化合物进行过滤,这些测试确定了它们在后期的特性,有效性和毒性。 越来越多地使用机器学习来更好地预测早期的分子特性,从而可以大大减少后续过程的负荷(例如临床试验),从而节省大量资源和时间。当前,机器学习在开发药物中的应用包括但不限于以下方面:通过广泛采用的定量结构-活性(性质)关系(QSAR / QSPR)模型进行生物活性或物理化学预测; 预测药物-蛋白质和药物-药物对的相互作用; 从头分子设计,产生具有所需药理特性的分子结构; 合成预测,预测合成反应的产物。由于传统的机器学习方法只能处理固定大小的输入,因此,大多数早期时代的药物发现都使用了特征工程,即生成和使用特定于问题的分子描述符。 通常,在这些任务中使用一组特定于问题的分子描述符作为特征。 常用的描述符包括:
- 分子指纹,通过一系列表示特定子结构存在的二进制数字来编码分子的结构;
- 由统计学家和化学信息学家处理的源自量子/物理化学和微分拓扑的描述符;
- 简化的分子输入行输入系统(SMILES)字符串,该字符串独特地描述了分子的结构并将其表示为行符号。 给定预定义的预测变量(即输入变量),然后通过机器学习算法构建分类或预测模型并进行学习。
近年来,越来越多的大型化学数据库可用于药物研究。因此,使用深层神经网络进行药物开发的新尝试已经出现。深度学习[1]取得了令人瞩目的成功,并在过去十年中被广泛用于自然语言处理[2]和计算机视觉[3]等领域的学习任务。深度学习的优势在于它能够从大规模数据中学习输入特征与输出决策之间的复杂关系。它在药物发现和分子信息学中的应用仍处于起步阶段,但已经显示出巨大的潜力。与毒品相关的工作中采用了几种常用的深度架构[4-7],与传统的机器学习方法相比,它们取得了实质性的进步。但是,由于以下原因,深层模型仍然存在局限性。首先,当前大多数深度模型仍基于手工特征或预定义的描述符,从而阻止直接从原始输入中学习结构信息。其次,现有的体系结构不适用于分子等结构化数据。在这些结构的特征提取过程中,既不考虑内部结构信息,也不充分利用结构信息。因此,更适合的架构对于进一步提高药物发现中深度学习的潜力至关重要。
结构化数据,例如图像,已经由卷积神经网络(CNN)成功地处理,卷积神经网络是深度神经网络的特殊体系结构。 CNN可以通过卷积运算符从原始图像中自动提取与任务相关的功能,从而揭示了图像相关任务的最新性能[8]。对于由原子和化学键组成的这类药物和小分子,我们具有不同的类型结构,即图,其中每个原子是一个节点,每个化学键是一个边缘。一个直接的尝试就是将卷积过程类似地应用于分子图。但是,与图像不同,图具有不规则的形状和大小。在节点上没有空间顺序,节点的邻居也与位置有关。因此,常规规则网格状结构上的传统卷积不能直接应用于图上。实际上,现实世界中广泛的结构数据通常以图而不是图像的形式形成,这意味着处理不规则结构的开发方法非常重要且迫切需要。
在整个文献中,人们都在努力对非欧几里德结构化数据上的卷积算符进行泛化,从而形成了所谓的图卷积网络(GCN)。 GCN已被确立为与毒品有关的任务的最新方法,其方式是:(1)通过考虑数据结构提取特征;(2)能够从原始输入而不是手工特征中自动提取特征可能会错过由于领域专家的偏见而导致的重要信息。当前出现的GCN遵循两个主要流。一个可以归纳为空间GCN,它通过对图中所有相邻节点的所有特征向量求和,直接在空间域中表示卷积。另一个称为频谱GCN,它根据频谱图理论在图谱域中定义卷积[9]。最近的工作[10,11]也表明频谱卷积可以被描述为空间卷积的一种特殊情况。但是,由于不同的理论基础,在以下各节中,我们仍将它们视为单独的卷积运算。在两个域中都定义了卷积后,生成型GCN利用卷积过程对隐藏的表示进行编码并生成分子图。
在本次调查中,我们特别关注GCN的最新进展及其在药物发现中的应用,而不是像以前的调查一样在一般深度学习的背景下进行介绍[12-14]。 因此,我们的综述重点是与药物相关的应用,包括最近的应用,旨在帮助读者深入了解新开发的药物发现深度架构的最新进展。 我们总结了Table. 1中最相关的论文。此外,我们还总结了这些研究中已使用的所有数据源,并在以后的章节中提供给公众使用。

其余内容安排如下:我们在图卷积原理一节中提供了图卷积的理论支持,并详细介绍了GCN的体系结构及其在GCN在药物发现中的应用方面的应用。 药物发现和分子生物信息学数据库部分概述了公共数据源的信息。 讨论部分讨论了当前方法之外的挑战和可能性。
Principles of graph convolution
Application of GCN on drug discovery
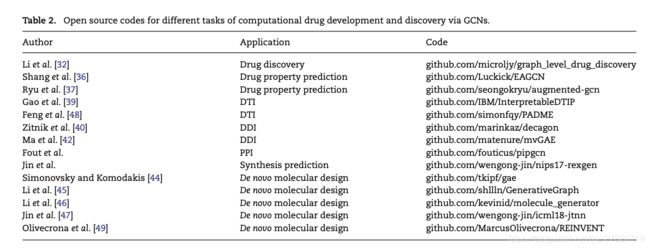
在本节中,我们回顾了先前有关计算药物开发和发现的主要应用的研究。 Table. 2列出了通过GCN进行计算药物开发和发现的不同任务的开源代码。
Quantitative Structure Activity/Property Relationship Prediction
QSAR(QSPR)可以预测生物学活性(化学性质)和分子描述符之间的关系。 关于这个问题的机器学习方法已经在文献中得到了广泛的探索[50-52]。 对于大多数与药物有关的计算方法,一个基本问题是要处理哪种类型的输入表示形式。 手工制作的特征无法完全编码分子图的结构信息。 此外,这些预定义的输入不是数据/任务驱动的,因此预测能力较低。 因此引入GCN来克服上述限制。
Biological property and activity
Duvenaud等人[28]首先提出了一种使用神经网络生成可区分且由数据驱动的指纹的方法。在这项研究中,规则的圆形指纹中的哈希函数(编码分子中每个原子的子结构)被替换为平滑函数。在这里,圆形指纹被设计为以不变于原子重新标记的方式编码分子中存在的亚结构[53]。因此,人们学会了用一个实值向量表示一个分子而不是一个二进制向量,这被称为神经指纹。分子的最终表示形式是通过汇总所有原子的表示形式而获得的,该表示形式穿过softmax层,该层能够解释所学特征。从聚集邻居信息以更新中心原子的意义上说,编码过程是卷积的,并且对具有相同邻居大小(范围为1至5)的原子及其相邻原子应用相同的局部过滤器。作者评估了所生成的指纹的几种药物特性,包括溶解度,药物功效和有机光伏效率,其中神经指纹优于传统的圆形指纹。而且,这些表示是可以解释的,使得与预测强烈相关的特征被分子结构中的某些片段所激活。但是,一个不足之处在于,与建立在预先计算的指纹之上的模型相比,训练指纹和预测模型都需要花费更多的时间,尤其是对于大型数据集。
除了节点特征或标签外,边缘信息也很重要,也可以在图卷积中进行编码。 Kearnes等人[29]提出了一种图形卷积框架来学习考虑节点和边缘特征的数据驱动任务的分子表示。具体来说,每一层都包含原子表示和成对(边缘)表示。所有关系模块的传播都跨越了不同的层:原子对原子(A→A),原子对对(A→P),对原子(P→A)和对对(P→P),形成一个编织模块。每层都遵循这种编织模块架构,而在最后一个卷积层,仅原子表示用于下游任务。通过神经网络可以实现跨相同表示(A→A,P→P)的转换。为了在不同表示(A→P,P→A)之间进行转换,在特征变换之后使用了额外的顺序不变的聚合操作。作者评估了259个数据集的生物活性方法,这些数据集由PCBA [54],Rohrer和Baumann构建的“最大无偏验证”数据集[55],有用诱饵的增强目录[56]和训练集Tox21挑战[57],在多任务环境中同时预测活动。输入是具有原子特征以及原子对特征的分子图。然后将建议的方法与使用RDKit生成的Morgan指纹的基线方法进行比较。拟议的WeaveNet并没有始终超越现有技术,但提供了一种除了节点功能之外还整合了边缘功能的方法。
Liu等人[33]也有类似的应用。 在这里,作者使用GCN开发了吸收,分布,代谢和排泄(ADME)属性预测系统。 卷积运算符类似于编织模块。 对于每个原子,首先通过完全连接的层转换邻域信息,然后使用不同的reduce运算符进行汇总和归约。 然后,通过将所有归约运算符(包括最大值,求和值和平均值)的结果进行级联来形成中心原子的表示形式。 然后将简化的表示形式与原子输入特征组合。 还针对五个选定的ADME终点对多任务方案进行了评估:人微粒体清除率,人CYP450抑制力,水平衡溶解度,孕烷X受体诱导和生物利用度,与基于规则的化学方法相比,拟议的Chemi-Net实现了改进 预测模型。为了获得分子表示,大多数先前的研究对原子水平表示进行求和或平均,但忽略了分子的图结构。 李等[32] 介绍了一种使用GCN生成图形级表示形式的替代方法。 作者通过引入虚拟超级节点(假定通过有向边连接到图中的所有节点)来实现其目标。 节点级别的传播遵循常规的空间图卷积。 对于虚拟超级节点,使用图及其本身中的所有节点(与之关联的其他权重矩阵)来更新表示。 作者评估了在生物活性分类和分子性质预测方面的建议方法。
先前的框架都是基于空间图卷积构建的,而频谱图卷积在QSAR(QSPR)任务中的使用较少,原因如下。 分子由大小不同的不同原子组成,因此最终具有不同的拓扑。 频谱GCN中派生的频谱受一个特定的图的影响,因此无法将其转移到其他图。 但是,仍存在关于在与药物相关的任务中使用光谱GCN绕过限制的研究。早期的光谱GCN固定图结构未经训练,因此无法从拓扑结构中学习。 李等[35] 构造的图卷积,它接受灵活的图输入并为每个输入图学习了其他拓扑信息。 图邻接矩阵通过参数距离度量更新,在训练过程中学习了用于调整距离度量的权重。 然后根据残差方案更新拉普拉斯算子,其中在每次迭代中,将学习的拉普拉斯算子的一小部分添加到原始拉普拉斯算子矩阵。 传播遵循常规频谱图卷积。 这种自适应设置允许输入具有唯一的拉普拉斯图,因此每个化合物都有其唯一的卷积滤波器。 作者通过在多个分子数据集上的多任务预测验证了该方法,并表明所学习的其他拓扑结构有助于提高预测精度。
除了节点特征外,即使对于同一分子,边缘属性也可能导致不同的图形表示。在以前的文献中还没有探索使用谱图卷积来共同学习边缘权重和节点特征。因此,尚等 [36]提出了一种基于边缘关注的图卷积网络来处理多关系图。在多关系图中,每个边缘特征(二进制或类别)都被视为一个关系。每个关系包含一个字典,其中包含每个关系类别要学习的值。该词典在所有图形之间共享,因此建议的方法对输入大小不敏感。卷积遵循一跳频谱卷积,在最后一层(预测)之前对不同关系的表示进行级联或加权平均。作者评估了四个不同数据集上的分类和回归任务。尽管由于字典设置的原因,所提出的方法适用于各种输入大小,但仅当边缘属性为二进制或分类变量时才可行。
同一原子通常根据其局部化学环境具有不同的分子特性。但是,先前的研究在不考虑其化学环境的情况下,对所有原子和键都具有同等重要性。为了解决这个问题,Ryu等人[37]引入了注意力机制来区分不同环境中的原子并提取决定分子性质的结构信息。给定一个中心节点,在聚合之前,每个邻居的注意力权重相乘。注意权重通过耦合矩阵表示中心节点和邻居节点来确定。传播遵循一跳频谱卷积。该网络包含六个卷积层和三个完全连接的层,并针对三个数据集进行了分子属性预测任务的验证。此外,作者还提供了可解释的综合结果,以比较建议的方法和一般的GCN。例如,在k均值聚类下对原子特征向量进行PC分析;两种方法对不同性质的分子之间的比较,表明注意力机制产生了更合理和可解释的结果。
Quantum mechanical property
除了生物学特性外,加快药物发现的另一个关键点是准确模拟分子动力学。 量子力学(QM)模拟,其中要扫描数百万个分子以确定其能量,对于预测分子的功效至关重要。 但是,传统的质量管理模拟方法,例如 用系统大小的 O ( N 3 ) \mathcal{O}\left(N^{3}\right) O(N3)进行缩放的密度泛函理论(DFT)方法非常昂贵,以至于它们只能适用于小型系统,或者采用精度较低的其他近似方法。 因此,一些研究集中于开发用于使用GCN快速筛选分子量子性质的深层结构。
Schutt等人[30]提出了一个用于预测分子总能量的深层张量网络。原子表示通过一系列交互过程进行细化,其中交互被定义为投影邻居表示和它们之间的投影距离的元素乘积的函数。然后,使用两个完全连接的层基于精确表示来计算每个原子的能量贡献。通过将所有原子的能量求和获得最终的分子能。后来,吉尔默等人。文献[10]进一步将先前的各种著作[21、22、28-30]重构为一个称为“消息传递神经网络”的通用框架,如空间卷积部分所述。作者提出了enn-s2s,这是消息传递网络的扩展变体,可以从分子图中提取特征。提出的框架使用键类型和原子间距离生成邻域消息,然后是用于将消息插入中心原子的set2set模型[58]。作者在QM9数据集上评估了他们的方法,该数据集由与分子相关的各种类型的能量和其他化学性质组成,并显示出优异的性能。
性能。
由于原子距离的离散化,在[10]中学习的滤波器也是离散的,无法捕获原子的逐渐位置变化并最终得到离散的能量预测。 Schutt等人[31]提出了另一个带有连续滤波器的图卷积方法,该方法将位置(距离)映射到相应的滤波器值。该体系结构由一系列原子层和交互层组成。原子层将特征映射重新组合为每个原子的新表示,并在原子之间共享权重,而交互层则根据原子距离更新了原子表示。在交互层中采用了残差连接,以便通过使用径向基函数作为连续滤波器生成器的卷积层获得残差。然后将所得的残差直接添加到原子表示中作为新的更新。作者通过预测三个不同数据集上的分子能量和原子力,证明了该方法的优势。
Incorporate GCN with other learning architecture
GCN最初是受传统卷积网络的启发,因此已有一些关于推广与传统CNN类似的GCN的研究。 Niepert等。 [27]在他们的工作中提出了这样的框架。面临的主要挑战是像传统CNN一样,为任意图形定义一个接收字段。作者首先通过使用图标记程序从图中选择了固定长度的节点序列来实现这一点。如果两个不同图中的节点在图中的结构角色相似,则将它们分配给相似的位置。给定选定的节点序列,然后通过广度优先搜索为每个节点组装邻域。之后,通过标准化组装后的邻域来构造节点的接受场。归一化过程旨在找到标记,以使矢量空间和图形空间中的两个图形之间的预期距离最小。每个顶点属性对应于一个输入通道。基于生成的接收字段,可以将任意功能或体系结构用于下行流任务。例如,作者在两个分子生物活性分类任务中使用了两个卷积层,然后是一个致密层和一个softmax层。
通过合并图卷积,最近还开展了利用内存网络对分子建模的工作。 在[38]中,Pham等人。 提出了用于对分子建模的图形存储网络。 存储器网络由控制器和外部存储器组成,其中存储器单元对每个节点的表示进行编码,并且控制器迭代地从存储器中读取和写入存储器。 图的表示是使用注意机制的所有存储单元的加权总和。 每个存储单元的表示均按照空间图卷积范例进行更新。 该控制器是使用跳过连接实现的。 由于通常在分子活性预测中的分子数目是有限的,为了呈现有限数据带来的过度拟合,作者使用了多任务方案来评估其方法。 共有9个生物测定活动测试,每个任务都有一个常数向量作为输入查询来表示不同的任务。
深度架构需要大量的训练数据,以实现对预测能力的显着改善,并且常见的是某些任务可能包含的数据不足,无法做出有意义的预测。通过将一次性学习与GCN结合起来,Altae-Tran等[34]证明,通过并入图CNN可以显着改善对小分子的合适距离度量的学习。目的是利用训练任务中的信息来构建测试任务的强大分类器,使用不同任务组中分子之间学习的表示形式的相似性,即查询分子的标签是基于支持分子的标签的加权总和。在他们的距离上。在提出的方法中,通过图卷积层获得分子表示。用于生成任务驱动的相似性度量的嵌入是通过迭代的长期短期记忆(LSTM)以残差净方式实现的。每次迭代时,都会通过细心的LSTM框架将一小部分添加到当前表示中。获得相似性度量后,可以立即得出分子标记的最终预测。作者在几个著名的分子特性数据集上评估了他们的方法。此外,通过将经过训练的模型转移到另一个数据集,作者表明,一次性模型在推广到其他不相关的系统方面的功能有限。
Interaction prediction
寻求用于疾病治疗的化合物的现代药物疗法依赖于几种类型的相互作用:
- 配体(可以是上市前化合物或现有药物的小分子)与蛋白质(靶标)之间的相互作用
- 蛋白质与蛋白质之间的相互作用在途径调控方法中准确定位相互作用的界面,
- 药物-药物相互作用(DDI),以检测潜在的不良反应并发现现有药物的新用途。
因此,相互作用预测在药物开发中也至关重要。但是,由于昂贵的实验分析以及在小型临床环境中药物和蛋白质之间复杂的相互作用的罕见性,这种相互作用的鉴定非常困难。根据当今的大型药物数据库,计算方法可在相对较短的时间内对潜在的相互作用进行大规模测试。传统的计算方法使用标准的机器学习算法来汇总药物和目标特征或相似性度量,以识别相互作用[59-62]。 GCN的出现使分子的可学习表示成为可能,从而提供了使用深度学习框架检测药物与靶标之间相互作用的新范例。
Ligand–protein (drug–target) interaction
预测配体(售前化合物或现有药物)与蛋白质(靶标)之间的相互作用是药物开发中的一个基本问题。 但是,由于以下原因,仍然存在一些挑战。 首先,可合成配体的化学空间是难以控制的,因此使预测仍然是一个未解决的问题。 其次,传统方法通常将交互作用预测视为一个二元分类问题,并且无法处理冷目标问题[63,64],即目标蛋白质从未出现在训练集中,这在实践中非常普遍。 第三,大多数传统方法虽然有效,但缺乏生物学解释,而在生物信息学领域却很重要。
因此,为了解决局限性,Feng等[48]提出了一个基于深度神经网络的框架,以预测化合物和蛋白质之间的实际值相互作用强度,而不是二进制类别标签。它的优越性取决于对带有GCN的每种药物的分子结构的了解,从而使药物表示具有更丰富的内部化学信息编码,而不是将每种药物仅当作知识图谱(KGs)中的一个节点。对药物的分子结构进行编码可以计算药物与蛋白质之间的实际值相互作用强度。这项研究以分子结构和蛋白质信息为输入,因此能够解决冷靶(和冷药)问题。 Lau和Dror [65]为晶体学数据开发了几种新颖的图卷积方法,表明通过图卷积学到的潜在特征在其他蛋白质-配体下游回归/分类应用中是有效的。
Gao等[39]提出了可以解释为预测药物与靶标相互作用的端到端深度框架。通过LSTM递归神经网络了解了由氨基酸序列组成的蛋白质的表达,同时通过图卷积层获得了药物分子的神经指纹[28]。引入了双向注意机制来跟踪药物原子与每个氨基酸成分相互作用的可能性,从而实现可解释性。然后使用注意力权重将原子聚合为分子表示(药物),将氨基酸聚合为蛋白质(靶标)。最后,将基于注意力的表示形式输入到分类器中进行预测。除氨基酸序列外,基因本体信息还用于推导蛋白质嵌入。可以将学习到的相互作用追溯到原子水平和氨基酸水平,以查看药物和靶标的哪一部分对相应的相互作用贡献最大。
显然,上述交互作用预测的性能取决于这些知识图的质量,即,这些KG中现有边的有效性。 但是,许多现实世界的知识图倾向于包含来自质量不同的多个来源的关系。 例如,从非结构化文本(例如医学文献)中提取的药物-靶标相互作用不如人工策划的可靠。 因此,必须有一种方法可以有效地利用噪声较大的便宜数据来进行更准确的预测。 为此,尼尔等人。 [66]通过增加网络的注意力参数来学习网络中的注意力参数,从而学习了GCNs的注意力机制,以在训练过程中信任边缘以减轻噪声边缘的影响。 它不仅可以提高干净数据集的性能,而且还可以很好地适应KG中的噪声。
Protein–protein interaction
尽管与药物设计没有直接关系,但是蛋白质-靶标相互作用为开发调节蛋白质途径的药物提供了靶标结合位点信息。 也可以使用图卷积网络预测此类交互。 在[67]中揭示了一个成功的应用程序。 给定两种蛋白质,每种蛋白质被馈入两层图卷积网络,并为每个原子学习嵌入。 然后通过原子嵌入的串联获得成对表示,然后是一个完全连接的层以对来自两种蛋白质的两个原子是否彼此反应进行分类。 实验中使用的特征是手工制作的,包括基于序列的特征和根据结构计算的特征。 因此,该应用程序旨在通过考虑繁殖步骤中的蛋白质结构,从现有特征中提取信息。
Drug–drug interaction
当两种药物同时使用而另一种药物影响一种药物的效果时,就会发生DDI。 DDI预测不仅有助于预防不良反应,而且还有助于发现新的药物用法(例如,有益的DDI可以为药物组合提供指导,并可以在治疗期间视为新药),从而提供有关药物开发过程的其他信息。因此,尽管DDI预测与狭义药物开发有间接关系,但我们仍将其纳入本节。 Zitnik等人[40]提出了一种基于GCN的框架,以进一步在多药副作用水平上识别DDI,即不同类型的DDI,为药物联合治疗提供了额外的指导。所提出的框架Decagon被描述为多模式网络中的多关系链接预测。它包含一个图卷积编码器和张量分解解码器。编码器考虑了具有两种类型实体的图形:药物和蛋白质。以及三种类型的相互作用:蛋白质-蛋白质相互作用(PPI),药物-蛋白质相互作用(DTI)和DDI。每个副作用都表示为不同类型的边缘。编码器遵循空间卷积约定,将不同的权重分配给具有不同关系类型的节点和邻居。每一层都被认为是一阶邻域,而更多的邻居则与堆叠层有关。编码器将每个节点映射到一个嵌入,而解码器旨在从学习的表示中重建边缘标签。在解码阶段,使用张量因数分解法为每个节点对计算关系得分。通过最小化交叉熵损失来优化整个模型。作者将他们的方法与张量因子分解模型和基于深度学习的方法(如DeepWalk [68])进行了比较,并取得了显着改进。
为了从异构数据源中获得更全面,准确的药物相似性,Ma等[42] 提出了一种使用带有注意机制的图自动编码器的多视图药物相似性集成框架。 每个视图都表示为由数据源中的要素获得的相似度矩阵。 通过注意机制汇总了不同的视图,在该机制中,每个视图都具有可学习的注意权重。 注意权重被对角线化以减少计算复杂度。 然后将融合的相似性内核输入到自动编码器框架中,以提取信息表示。 传播遵循谱图卷积,如[22]。 作者评估了针对DDI发生和具有不同关系类型的DDI的DDI识别任务的建议方法。
结构信息也可以与其他数据类型组合以增强预测性能。 浅田等[41] 提出了从文本中提取DDI的框架。 给定一个带有药物提及的语料库,目标是将药物-药物对分类为不同类型的相互作用(机理,作用,建议,内在相互作用和无相互作用)。 传统方法使用CNN提取特征以预测药物-药物关系[69]。 作者首先通过预先训练用于DDI识别的GCN(二进制)将药物结构信息纳入框架。 之后,将来自固定GCN的药物嵌入与来自CNN的文本嵌入连接起来,送入一个完全连接的层中以预测相互作用类型。 药物结构来自DrugBank,文本文档来自DDIExtraction 2013共享任务[70]。 结果表明,结构信息可用于从文本中提取DDI。
Synthesis prediction
预测有机反应结果是设计产生特定目标分子的反应序列的基本步骤。它涉及两个步骤:候选人生成和筛选。候选生成的最新解决方案基于反应模板,该模板指定了可以应用的分子子图模式以及相应的图转换。模板是手工制作或从反应数据库生成的[71-73],该数据库存在覆盖范围和效率问题。而且,匹配过程是昂贵的,使得当前的方法仅适用于小的数据集和有限的反应类型。在[43]中,Jin等。提出了一种无模板方法,用于利用图神经网络进行反应预测。输入是预定义的原子和键特征。利用图卷积,网络学会了通过预测反应物分子中每个原子对的反应性得分而无需使用模板即可识别反应中心。前向传播遵循空间图卷积。得分最高的原子对用于生成候选产品。然后建立了第二个网络,用于对候选人进行排名,以找到真实的反应结果。作者提出了两种用于候选人生成和排名阶段的模型。在模型生成的候选集中找到真实产物的反应比例用作评估反应中心的评估指标。覆盖精度用于候选者排名。结果表明,所提出的方法在很大程度上优于基于模板的方法。
在化学领域,逆合成是设计化合物生产的标准方法。原理是,从精神上往后看,化合物会分解成越来越小的成分,直到获得基本成分为止。该分析提供了“烹饪食谱”,然后将其用于实验室中,从原材料开始生产目标分子。尽管从理论上讲很容易,但是该过程在实践中存在困难。就像在国际象棋中一样,您在每一个步骤或动作中都有多种选择。但是,在化学中,比象棋有更多数量级的可能动作,而且问题要复杂得多。计算机辅助的逆向合成将是非常有价值的工具。但是,过去的方法很慢,并且结果不令人满意。为此,Segler等人[74]提出了一种基于深度学习的新的计算机辅助合成计划(CASP)方法,该方法利用蒙特卡罗树搜索有效地发现逆合成途径,现在着重于将发现付诸行动。与传统的CASP方法相比,该新方法在很大程度上借鉴了深度神经网络和强化学习的思想,并且是对传统CASP方法的重要改进。与计算机科学界基于GCN的方法相比,Segler等人提出了这种新方法的设计。借用了很多化学知识,反映了对化学反应的深刻理解。
De novo molecular design
药物开发的最终目标是发现具有所需药理特性的新化学结构。然而,由于实际上无限的搜索空间,实际上药物设计是困难且昂贵的[75]。因此,从头分子设计旨在利用计算方法来自动化分子生成过程。早期的研究利用基于规则的方法来减少搜索空间并生成分子[76,77]。深度学习中的生成模型可以基于SMILES字符串有效地生成分子[49,78]。但是,SMILES和指纹太简单,无法提供分子结构的拓扑信息,并且导致学习准确性相对较低。分子图可以直观,简明地表达具有2D拓扑信息的分子。因此,它们在化学教育以及化学信息学中被广泛采用。实际上,已经进行了努力来开发基于分子图的DL模型。作为CNN的扩展,GCN引入了分子的新表示形式,因此可以直接实现生成分子图,而不是通过管道实现方式生成中间表示,从而为分子生成提供了新的思路。 GCN受益于CNN架构的优势;与完全连接的多层感知器模型相比,它利用较少的参数以较高的精度执行,但计算成本相对较低。它还可以通过分析相邻原子之间的关系来确定重要的原子特征,这些特征决定了分子的性质。分子图中相邻原子之间的信息传播设计是对分子中结构信息进行编码的简单有效方法[28]。
Simonovsky等人[44]提出了用于生成小分子图的变分自动编码器。编码器由变分后验定义,解码器由生成分布定义,每个都包含可学习的参数。编码器的输入是图邻接矩阵,边缘特征张量和节点特征。作者使用边缘条件卷积作为编码器。解码器在预定义数量的节点上输出概率完全连接图,可以从中提取离散样本。通过最小化负对数可能性的上限来训练模型。通过近似图匹配来对齐生成的图和地面真相,可以增强自动编码器的重构能力。作者将基于GCN的VAE与传统的基于字符的生成器[79]和基于语法的生成器[80]进行了比较,并论证了提出的生成化学上更有效的分子的方法。但是,由于其预定义的节点数始终等于或大于实际分子大小,因此提出的模型仅对生成小图有用。而且,输出是一个密集的表示,使得参数和匹配的复杂性迅速增长。
Li等人[45]研究了一种概率方法,该方法顺序生成原子并扩展图,而不是立即生成整个图。生成过程可以视为关于添加节点还是边缘并选择一个节点与新节点连接的一系列决策。决策是根据GCN描绘的概率做出的。通过最大化图上预期的联合对数似然分布以及其节点和边的排序来学习网络。生成模型可用于实现条件生成。典型的输入用于调节生成过程。作者评估了所提出的方法,用于生成特定拓扑图和分子图。结果表明,与具有LSTM体系结构的模型相比,该方法可生成更有效的图形。但是,可伸缩性仍然是所提出方法的挑战,因为大型图需要更多的传播步骤来确保信息流,并且训练这种图模型比训练LSTM网络更加困难。 Li等人[46]采用了类似的框架。在每个步骤中,都会采样并执行图转换(追加,连接,终止)。使用GCN对每个过渡采样的概率进行参数设置。过渡映射被表示为解码方案。作者探索了两种解码策略,一种是通过马尔可夫过程参数化的,另一种是使用分子水平的递归单元来增加模型的可扩展性。
以原子方式生成分子图的主要缺点是产生低质量的中间体。 Jin等[47]提出了一种利用子结构作为有效成分的两阶段分子图生成方法,从而显着提高了生成分子的质量。该方法首先生成一个连接树结构来表示子图组件,这些子图组件用作构建块。在第二阶段,将子图组合在一起成为分子图。该图由标准GCN编码。通过平均所有节点表示获得最终的图形表示。使用消息传递网络对树结构进行编码,其中消息是通过门控循环单元构造的。最终的树表示形式是从树根编码的。在解码过程中,首先对一棵树进行解码,然后通过对子图进行枚举和排序来从连接树中对图进行解码。作者通过三种方式对提出的方法进行了评估:
- 分子重构和有效性:从其潜在表示中重构输入分子,并在从先验分布采样时对有效分子进行解码
- 贝叶斯优化:测试模型如何产生具有所需特性的新型分子
- 约束分子最优化:修饰给定的分子以改善特定的特性,同时约束与原始分子的偏离程度。将该方法与基于SMILES的最新VAE进行了比较[80,81],并显示出显着的改进。
Databases for drug discovery and molecular bioinformatics
到目前为止,我们已经介绍了GCN在药物发现中的应用。 在本节中,我们在Table. 3中提供了被调查论文所使用的数据库的摘要。 我们将它们分为与药物应用相对应的组。 我们还包括多个数据库,这些数据库是从多个来源集成而来的,可用于开放访问。


Molecular property and activity
Biological property and activity
PubChem [54]是一个大型公共数据库,提供化学分子及其对抗生物测定的活性。 它由三个主要通道组成:PubChem生物测定(PCBA),PubChem化合物和PubChem物质。 物质包括化合物及其所有参与者报告的信息。 化合物衍生自物质,由该物质的化学结构的标准化表示组成。 BioAssay包含来自125万个高通量筛选程序的生物活性结果。 PubChem化合物编号通常在不同的化学数据库中使用,以引用相同的化合物。
最大无偏验证(MUV)[55]数据集是PCBA的子集,它是使用精确的最近邻分析产生的,并且在模拟偏倚和人工富集方面没有偏见。 它包含约9万种化合物的17项艰巨任务,是专门为验证虚拟筛选技术而设计的。 选择这些数据集中的阳性示例在结构上彼此不同。
ChEMBL [82]是具有类似药物性质的生物活性分子的数据库。 它包含分子化合物的结合,功能和ADMET(吸收,分布,代谢,排泄和毒性)信息,这些信息是手动从主要已发表的文献中获得的,然后进行进一步的标准化。 该数据库提供了540万种生物活性测量结果,涉及超过100万种化合物和5200种蛋白质目标。
ZINC [83,84]包含精选的为虚拟筛选而制备的市售化合物。 它从20多种资源中提供与分子相关的信息,例如化学结构,生物活性和目标信息。 它包含了超过2亿个即插即用的3D格式的化合物。
NCI [85]是针对针对不同癌细胞系的活性进行筛选的化学化合物数据库。 它包括约250K分子的生物学测试数据和化学结构。 由药物治疗计划[86]使用AIDS抗病毒筛选产生的HIV数据集,也是NCI数据库的一部分,在该数据库中检查了化合物的抗HIV活性证据。 对筛选结果进行了评估,并将其分为三类:已确认活跃,已确认无效和已确认中等活跃。 该数据集包含43850种化合物的筛选结果以及结构信息。
Tox21 [57],ToxCast [87]和ClinTox [63]是包含分子毒性信息的数据集。 Tox21,在21世纪计划中被称为毒理学,是联邦机构之间的合作,旨在开发创新的测试方法,以更好地预测物质如何影响人类和环境。 它包含对12个生物目标的定性毒性测量,包括核受体和应激反应途径。 ToxCast是由环境保护署提供的数据集,用于开发有效的方法来对化学品进行优先级排序,筛选和评估。 它使用高通量筛选方法和计算毒理学方法,包含来自多种来源的1800种化学物质的毒理学数据。 ClinTox是MoleculeNet [63]基准数据的一部分,其中包括因毒性原因而在临床试验中失败的药物化合物以及FDA批准的那些化合物。 它包含1491种药物化合物的两个分类任务。
FreeSolv [88]是实验和计算出的水中小分子水合自由能的数据库。 这些值是使用分子动力学模拟从炼金术自由能计算得出的。 它目前包含643个分子的分子性质数据及其化学结构。 ESOL [89]是另一个包含数以千计的低分子量化合物的均一溶解度数据的数据集。
Quantum chemical property
清洁能源项目数据库(CEPDB)[90]是哈佛清洁能源项目的数据库,这是一项虚拟的高通量筛选计划,旨在确定有前途的碳基太阳能电池材料新候选人。 该项目建立了一个自动化的计算机硅框架,以研究有机光伏的潜在候选结构。 该数据库提供了从DFT模拟获得的有关230万个候选分子基序的信息,这些基序包括已知化合物和虚拟化合物。
量子机器(QM)[30,91–96]是一个包含分子及其量子力学特性(例如原子能和力)的数据库。 它包含来自多个来源的数据,旨在加速机器的开发,该机器可以根据第一原理快速准确地模拟量子化学系统。 自2013年以来,它已经发布了QM7,QM8,QM9和MD数据集。
Interaction database
副作用资源(SIDER)[97]是一个数据库,其中包含有关上市药物及其记录的药物不良反应(ADR)的信息。 该信息是从公共文档和包装说明书中提取的。 当前,数据库中有1430种药物和5868种副作用(SE),其中有139756种药物-SE对。 还提供了药物适应症的数据集以减少假阳性。
标签外副作用(OFFSIDES)[98]是针对1332种药物和10097种不良事件的438801标签外副作用的数据库。 标签外的副作用表示未在FDA官方药品标签上列出的副作用。 使用不良事件报告系统收集信息,该系统收集患者,医生和公司的报告。 由同一实验室生成的TWOSIDES [98]是药物对多药房副作用的一种资源。 它仅包含药物组合引起的副作用,而不是任何单一药物引起的副作用。 与OFFSIDES一样,信息是通过不良事件报告系统生成的。 该数据库包含59 220对药物与1301不良事件之间的868 221个重要关联。
相互作用化学品搜索工具(STITCH)[99]是一个数据库,它将430万种化学品和960万多种蛋白质的数据源整合到一个资源中。 它提供了化学物和不同相互作用目标之间的结合亲和力,从而形成了化学-化学相互作用和化学-蛋白质相互作用的全球网络。 蛋白质空间与蛋白质网络中的蛋白质网络STRING [100]共享。 所有交互都与代表网络实体之间链路强度的置信度得分相关。
DrugBank [101,102]是一个全面的数据库,其中包含有关药物的详细分子信息。 数据库中有两种药物,FDA批准的小分子药物和生物技术药物。 它还提供有关目标,指标和途径的信息。 每种药物的数据字段都与其他数据库(PubChem,ChEBI,PDB,KEGG等)超链接。 最新版本包含11680个药物条目和5129个与这些药物条目链接的非冗余蛋白(即药物靶标/酶/转运蛋白/载体)序列。
治疗靶标数据库(TTD)[103-106]包含有关文献中描述的已知治疗蛋白和核酸靶标的信息。 除目标外,它还提供相应药物的信息,目标疾病状况和途径信息。 序列和结构信息也可以通过与其他数据库的交叉链接获得。 目前,数据库包含3101个目标和34 019种药物。
对接基准数据库(DBD5)[107]是一个基准数据库,其中包含一组不同的蛋白质对接测试用例。 复合物是蛋白质数据库(PDB)中结构的子集选择。 它包括单独结晶的受体和配体PDB,以及共结晶的复杂PDB,用于测试蛋白质对接算法。
BindingDB [108]是在药物靶标蛋白质和小的药物样分子相互作用中测得的结合亲和力的数据库。 该数据库包含1 454 892结合数据,其中包含7082个蛋白质靶标和652068个小分子。
Synthesis database
USPTO [109]是一个数据库,其中包含化学反应物的反应信息。 这些反应摘自美国专利商标局(USPTO)的专利申请。 总共提取了424621个精确的原子映射反应,并将其包括在数据库中。
Integrated benchmark database
MoleculeNet [63]是旨在测试分子性质的机器学习方法的基准。 它建立在多个公共数据库的基础上,涵盖了700,000种经过一系列不同性能测试的化合物。 数据集分为四个类别:生理学,生物物理学,物理化学和量子力学。 它包含上述数据库的子集合,包括QM,毒性数据集(Tox21,ToxCast。ClinTox),生物活性数据集(PCBA,MUV,HIV),生物属性数据集(ESOL,FreeSolv)和相互作用数据集(SIDER)。 它还包含其他数据集,例如BBBP,血脑屏障渗透的二进制标签; 亲脂性,辛醇/水分配系数的实验结果; BACE,一组人β-分泌酶1抑制剂的结合结果; 和PDBbind,对生物分子复合物的结合亲和力。
Decagon [40]提供了蛋白质靶标和药物分子及其相互作用的预处理数据。 该网络由DTI,PPI和DDI组成,这些DTI,PPI和DDI来自多个数据源。 通常,DTI是从STITCH数据库中提取的; DDI来自TWOSIDES数据库; PPI是通过人工PPI网络和STRING数据库进行集成的。 还包括SIDER和OFFSIDES的药物副作用。
为了提高从头分子设计生成模型的评估的一致性,Brown等人。 [110]基于一套标准化基准引入了一种评估框架GuacaMol。 基准任务包括测量模型的保真度以重现训练集的属性分布,生成新分子的能力,化学空间的探索和开发以及各种单目标和多目标优化任务。 基准测试框架可作为开源Python软件包获得。
Polykovskiy等。 [111]介绍了用于药物发现的MOSES,它实现了几种流行的分子生成模型,并包括一组评估生成的分子的多样性和质量的指标。 MOSES旨在使分子生成研究标准化,并促进新模型的共享和比较。 此外,它对现有的最新模型进行了大规模比较,并详细阐述了生成模型当前面临的挑战,这些挑战可能为新研究提供沃土。 该平台应允许公平,全面地比较新的生成模型。
Discussion
正如最近的一篇综述文章所总结的那样,GCN可以被描述为在对结构化数据进行建模时施加了相关的归纳偏差[112]。 GCN的兴起及其在分子生物信息学等领域的成功应用说明了结合深度学习(假定先验知识最少)和结构化方法(对输入和模型施加严格约束)的强大功能。从广义上讲,GCN适用于可以表示为图形的任何数据结构,因此在各种实际应用中具有巨大的意义。
在药物发现领域,深度学习能够在较短的时间内对化学性质和活性进行大规模预测,从而自动化并加快药物发现过程。通过自然地考虑分子结构,与传统方法相比,图卷积网络的引入提供了更准确的预测。此外,当与其他机制(例如注意力)结合使用时,图卷积网络会在交互预测中生成生物学可解释的结果。
但是,尽管图卷积网络最近取得了成功,但要想完全释放图卷积网络在药物发现方面的潜力,仍然存在挑战。在此,我们在以下小节中总结了挑战和机遇。
Database challenges and opportunities
深度模型需要大量数据才能了解输入和目标之间的复杂关系。尽管可以使用大型数据库,但是由于以下原因,仍然存在功能不足的问题。首先,对于某些分子特性(例如溶解性,毒性),可用数据有限或作为不同的小型数据集传播。除了收集更多数据外,一个更好地集成不同数据源的统一平台对于交叉引用和获取更多数据也至关重要。其次,现有数据库主要收集阳性样本。例如,在交互网络中,如果两个实体进行交互,则将它们包含在数据库中,而通常忽略非交互对的信息。这不仅会引起不平衡问题,还会导致完全无法获得控制信息的情况。当前的计算方法要么设计新目标,要么手动生成负样本以克服局限性,而实际上很难确定负样本。因此,官方策划的负样本对于使用机器学习方法进行更准确的预测非常重要。第三,可以将更多指定的详细信息添加到数据库中。例如,即使对于交互对,交互也可能具有不同的功能。当一种药物与另一种药物相互作用时,其作用可能是协同作用或拮抗作用,而实际上仅记录了拮抗作用[113]。实际上,DDI的协同作用是有益的,因此可以为患者护理中的药物组合提供重要指导[114]。
Methodology challenges and opportunities
分子化合物,尤其是蛋白质,是3D形的实体,在3D空间中的折叠结构会极大地影响其功能[115]。当前的图卷积主要在平面二维图上运行,而二维空间中的结构信息被忽略。已经有一些尝试在3D结构上开发卷积算子[116-118],并将图卷积网络扩展到3D结构绝对是一个值得探索的方向。
另一方面,虽然高阶结构实际上可以提供其他信息,但它们对二维图的关注较少,而在二维图上却很少。例如,在疾病-蛋白质网络分析中[119],作者发现疾病路径并不对应于单个良好连接的组件,而更高阶的网络结构(基序)为疾病路径发现提供了更多信息。 Monti等。 [120]研究了谱图卷积的图案,并在引用网络数据上得到了验证。基于基序的图卷积的探索及其在药物发现中的应用尚未得到很好的建立,因此是一个有前途的未来方向。
现有图卷积在规则图上运行,而对于某些关系,可以形成超图。例如,不同的药物可能共享相同的ADR,目标或适应症,可以转换成超图。文献中尚未研究如何在超图上定义适当的卷积以提取有用的信息。
Network design challenges and opportunities
当前,图卷积网络通常用于两种情况。在第一种情况下,每个数据点都表示为图,其中预测发生在图级别,例如分子性质和活性预测。在第二种情况下,仅呈现一个图形,并且每个数据点表示图形中的一个节点,例如药物-靶标相互作用网络。第一个旨在提取每个实体的结构信息,而第二个旨在在实体之间传播亲和力信息。
可以做的一项改进是结合两个场景,并利用底层结构信息和全局网络结构信息来构建端到端框架。对于第二种情况,通常在交互网络中最多显示两个实体,而实际上,药物发现涉及两个以上的实体,其他实体有助于提供其他信息。例如,在更大的网络中,实体可以是药物,目标,疾病甚至是ADR(关系)。但是,在文献中没有很好地考虑具有两个以上模态的多模态网络的图卷积。造成这种情况的一个可能原因是,尽管图卷积网络适用于可以表示为图的任何数据,但是,图表示并不总是针对现有数据。例如,分子可以自然地成为图形,而病历则不是,尽管可以精心设计以形成某些图形。在用于药物发现的多模型网络的情况下,具有三种或更多类型的实体通常更复杂,因此设计适当的图以应用卷积框架至关重要。
Interpretability challenges and opportunities
由于深度神经网络的复杂性,它总是会受到缺乏可解释性的批评。 但是,在生物信息学和与健康有关的领域中,可评估性在评估计算模型和更好地理解潜在机制时非常重要。 因此,设计能够解释或可视化复杂关系的微妙架构既是挑战,也是药物研发中GCN应用的机遇。 先前的研究[39,67]成功地使用注意力机制或节点对评分显示了药物和蛋白质实体(DTI和PPI)之间的相互作用复合体。 需要其他机制来进一步提高学习模型的可解释性。
Key Points
- GCN是一类计算技术,旨在通过图卷积从通用图中提取特征。
- 如果我们将每个药物分子结构视为一个以原子为结点,键为边的图形,则GCN可以应用于药物开发。
- GCN已成功应用于许多药物开发问题,包括QSAR / QSPR,药物靶标/ DDI预测和Denovo药物分子结构设计。
- 有许多可公开获得的药物相关数据库,用于在各种计算药物开发应用程序中开发基于GCN的方法。
- GCN在计算药物开发方面仍然面临挑战,包括全面的数据,优化的模型设计和模型的可解释性。