NBT封面:水稻NRT1.1B基因调控根系微生物组参与氮利用

https://www.nature.com/nbt/volumes/37/issues/6
Nature Biotechnology 杂志2019年6月5日(37卷第6期),中科院遗传发育所关于水稻微生物组的研究入选为封面文章。

原文链接:https://www.nature.com/articles/s41587-019-0104-4
官方分享链接(免费阅读PDF全文):https://rdcu.be/bFHLH
相关报导
官方新闻稿
关于文章的科普性介绍和新闻稿,请点击访问下方链接
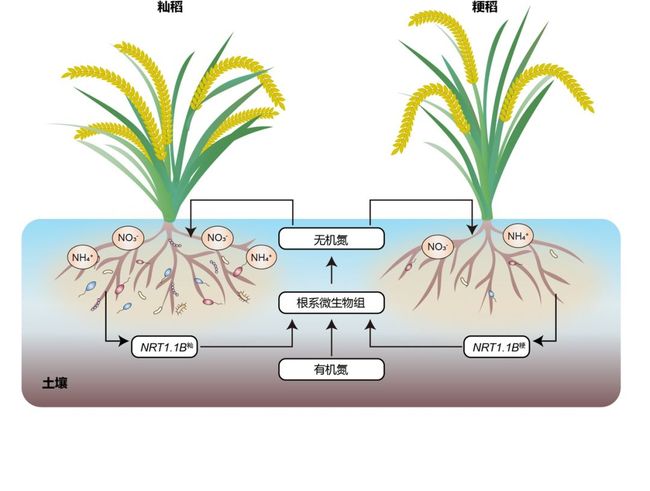
Nat. Biotechnol.|中科院揭示籼粳稻根系微生物组与氮肥利用效率关系
植物学报评论
关于王二涛研究员在植物学报配发关于本文重要性的专题评论,请阅读

·热点评· 根际微生物促进水稻氮利用的机制
热心肠日报导读
第1088期封面文章:食不可无大米,稻不可无菌群!中科院新成果登上NBT
https://www.mr-gut.cn/papers/read/1070836774
中科院:揭示籼粳稻根系微生物组与氮肥利用效率的关系
创作:刘永鑫 审核:刘永鑫 04月30日
原标题:水稻NRT1.1B基因调控根系微生物组成并参与氮利用
亚洲栽培稻主要分为籼稻和粳稻两个亚种,籼稻通常表现出更高的氮肥利用效率;
籼稻和粳稻形成了截然不同的根系微生物组,可以作为区分籼粳稻的生物标志;
籼稻根系比粳稻富集更多与氮循环相关的微生物种类,具有更加活跃的氮转化环境;
水稻通过NRT1.1B调控根系具有氮转化能力的微生物,改变根际微环境,影响籼梗稻田间氮肥利用效率;
通过高通量微生物分离培养,获得了水稻根系70%的细菌种类,建立了首个系统性水稻根系细菌资源库。
主编评语:中科院遗传发育所白洋和储成才组近期在《Nature Biotechnology》发表了关于水稻微生物组的成果,该项研究不仅揭示了水稻亚种间根系微生物组与其氮肥利用效率的关系,证明了NRT1.1B在调控水稻根系微生物组的关键作用,还建立了第一个水稻根系可培养的细菌资源库,为研究根系微生物组与水稻互作及功能,为应用有益微生物、减少氮肥的施用奠定了基础。
水稻NRT1.1B基因调控根系微生物组成参与氮利用
*NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice
Nature Biotechnology [IF:35.724]
在线时间:2019年4月29日 23点
出版时间:2019年6月4日 23点
DOI:https://doi.org/10.1038/s41587-019-0104-4
第一作者:张婧赢1,2,10、刘永鑫1,2,10、张娜1,2,3,10、胡斌1,10、金桃4,5,6,10
通讯作者:储成才1,3([email protected])、白洋1,2,3([email protected])
其它作者:徐浩然、秦媛、阎鹏旭、张小宁、郭晓璇、Jing Hui、曹守云、王鑫、王超、汪辉、曲宝原、Guangyi Fan、袁立行、Ruben Garrido-Oter
研究单位
中国科学院,种子创新研究院,遗传与发育生物学研究所,植物基因组学国家重点实验室 (State Key Laboratory of Plant Genomics, Institute of Genetics and Developmental Biology, The Innovative Academy of Seed Design, Chinese Academy of Sciences, Beijing, China)
中国科学院遗传与发育生物学研究所,植物与微生物科学中英联合研究中心(CAS-JIC Centre of Excellence for Plant and Microbial Science, Institute of Genetics and Developmental Biology, Chinese
Academy of Sciences, Beijing, China)中国科学院大学,现代农学院(3College of Advanced Agricultural Sciences, University of Chinese Academy of Sciences, Beijing, China)
深圳华大基因研究院(BGI-Shenzhen,
Shenzhen, China.)青岛华大基因研究院(BGI-Qingdao, BGI-Shenzhen, Qingdao, China)
深圳华大基因,国家基因库(China National Genebank-Shenzhen, BGI-Shenzhen, Shenzhen, China)
中国农业大学,资源与环境学院,植物-土壤相互作用教育部重点实验室(Key
Lab of Plant–Soil Interactions, MOE, College of Resources and Environmental Sciences, China Agricultural University, Beijing, China)德国科隆,马克思普朗克植物育种研究所,植物微生物互作中心(Department of Plant
Microbe Interactions, Max Planck Institute for Plant Breeding Research, Cologne, Germany)德国杜塞尔多夫,植物科学卓越创新中心(Cluster of Excellence on Plant Sciences, Dusseldorf, Germany)
共同第一作者
摘要
模式植物和作物的根系微生物组已经得到了广泛研究,但与在植物群体层面根系微生物组的变异,以及与遗传因素的联系仍不清楚。
水稻籼稻(indica)品种比粳稻(japonica)有更高的氮利用效率。我们在水稻的两个主要亚种(包括68个籼稻和27个粳稻品种)为研究材料,发现在田间籼粳稻招募不同的根系微生物组。籼稻比粳稻有更高的微生物多样性,其中包括更多氮代谢相关的属。应用机器学习方法挖掘的生物标记,可以很好通过根系微生物组区分籼粳稻亚种。
进一步分析nrt1.1b基因突变体和近等基因系,发现水稻的氮转运和受体NRT1.1B基因与大部分籼稻富集菌有关。比较nrt1.1b基因突变体和野生型的根系宏基因组数据,发现氨化过程相关基因丰度明显下降。我们采用梯度稀释方法培养结合标签高通量测序技术,从水稻籼、粳稻根系分离鉴定了1079株细菌纯培养。因此可以设计人工重组群落接种于无菌生长的籼稻品种IR24,验证了籼稻富含菌比粳稻富集菌具有明显的生长促进作用。
本研究发现了植物表型与根系微生物组成员的关系,可用于进一步开发育种策略,改进作物品系的氮利用效率。
编辑总结:水稻与根系微生物组合作改善土壤中氮素获取
Rice coordinates recruitment of the root microbiota to optimize nitrogen acquisition from soil
背景
植物多种性状的进化与土壤环境中丰富多样的微生物相关。许多微生物参与土壤养分的生化转化,包括植物生长必须的氮和磷元素循环。在模式植物和作物中,植物根系从土壤中选择性的招募根系微生物。己有研究报导根系微生物的决定因素,包括根不同区域(compartment)、土壤类型、地理位置、营养状态、发育阶段和宿主基因型。然而在植物群体水平遗传因素对根系微生物组的影响至今仍不清楚。
水稻是世界范围内最广泛种植的主食。亚洲地区的两个栽培水稻亚种:籼稻、粳稻被大家广为熟知。籼、粳稻在基因组和表型层面的差异已经有非常详细的文献报导。在田间条件下,与粳稻相比,籼稻一个显著的表型差异是更高的氮利用效率(nitrogen-use efficiency, NUE)。关于调控氮利用效率的基因,有报导氮转运和受体基因NRT1.1B的自然变异(第327位氨基酸:籼稻为蛋氨酸Met,而粳稻为苏氨酸Thr),可部分解释籼粳稻的氮利用效率差异。土壤中氮中主要形式为氨盐、硝酸盐和有机氮。因为植物相比有机氮更容易吸收无机氮的铵盐和硝酸盐,土壤传播的细菌可以代谢不同形式的氮,可能影响植物氮的吸收。因此提出科学问题,植物根系微生物组是否参与植物不同亚种如籼粳稻间的氮吸收效率不同。此外,水稻根际微生物处于长期淹水的环境下形成了特殊的根系微生物组。之前的研究只报导了水稻2-4个品种基因差异对水稻根系微生物组的影响。然而,至今没有水稻群体水平研究间微生物组差异的研究。目前有丰富的水稻亚种资源,可以在群体层面开展水稻基因与微生物组的关联研究,挖掘水稻驯化过程中调控根系微生物组的关键基因。
本研究中,我们分析了种植在田间的68个籼稻品种和27个粳稻品种的根系微生物组,鉴定了根系微生物组、土壤氮循环、宿主遗传调控和氮利用间的关系。
结果
籼粳稻存在不同的根系微生物组
Indica and japonica have distinct root microbiota
为探索栽培水稻的根系微生物组,我们在海南陵水农场两个不同地块种植了68个籼稻品种和27个粳稻品种(附表1. 两块地的土壤属性和耕作历史)。这些品种来自美国农业部水稻种质库的微核心资源,来源包括了44个国家和地区(图1a;附表2. 籼粳稻品种信息)。稀释曲线分析样本量后期进入平滑期表明我们的品种资源已经足够包括各亚种的绝大多数根细微生物组种类(附图1)。为避免种子来源的微生物引起污染,我们首先对种子灭菌,然后在MS培养上生长15天,再移植于田间,各品种的种植位置进行随机排布(图1b)。这两地块在过去10年一直种植水稻,在2014-2016年存在明显不同的耕作方式(附表1;在线方法),这样的设计有利于我们研究田间不同种植方式下水稻根系微生物组的稳定性。取样时间为水稻转移至土壤中8周,这一阶段可形成较稳定的水稻根系微生物组。对于每个水稻品种,我们从三个独立生物学重复的株取分别取1个样本(图1b)。在每个地块收集12个末种植土壤样本(Bulk soil)。采用PCR扩增细菌16S rDNA的V5-V7区(引物799F和 1193R)并进行Illumina测序。共获得了563个样本,包括19,976,393条高质量的序列,平均35,482条序列; 每个样本测序量范围: 5,386–125,709条序列。我们采用USEARCH进行分析移除嵌合体系,SILVA数据库比对去除细胞器序列,产生5,141个OTUs(平均每个样本包括1,455个OTUs;附表3包括OTU表、代表序列和物种注释;原始序列储存于北京基因组所(BIG)大数据中心的基因组序列存档(GSA)中,访问编号:CRA001372)。
我们发现不同水稻亚种根系微生物组成存在差异。基于Bray-Curtis距离的非限制性主坐标轴分析(PCoA)表明籼粳稻在地块1的微生物组成明显形成两大类,且在第一主轴分开(图1c;附图2),表明水稻亚种分化是微生物组变异的最主要影响因素。同时也观察到了由于地块2的土壤不同,在地块2存在微生物组的变化(附图3;附表1)。但在两块地块中,籼粳稻显著分开保持一致(图1d;附图3)。籼粳稻的alpha多样性也存在显著差异(图1e;附图4),籼粳的多样性显著高于粳稻(图1e),表明籼稻的根系比粳稻招募了更多种类的微生物。籼粳稻间微生物组差异在门水平也存在显著差异(图1f)。在两块地中,籼、粳稻间根系微生物组相比,籼稻更富集δ-变形菌纲(Deltaproteobacteria)、放线菌门(Actinobacteria)、产酸菌门(Acidobacteria)、拟杆菌门(Bacteroidetes)、螺旋体门(Spirochaetes)、绿弯菌门(Chloroflexi)、 Ignavibacteriae、硝化螺旋菌门(Nitrospirae)和疣微菌门(Verrucomicrobia),而粳稻更富集α-变形杆菌纲(Alphaproteobacteria)(Wilcoxon秩和检验,FDR < 0.05,附表4. 籼粳稻间差异的门和变形菌纲)。这些数据表明,水稻亚种间差异的微生物组,在不同地块中可重复。
图1. 籼粳稻的根系微生物组
Fig. 1 | Root microbiota of indica and japonica.
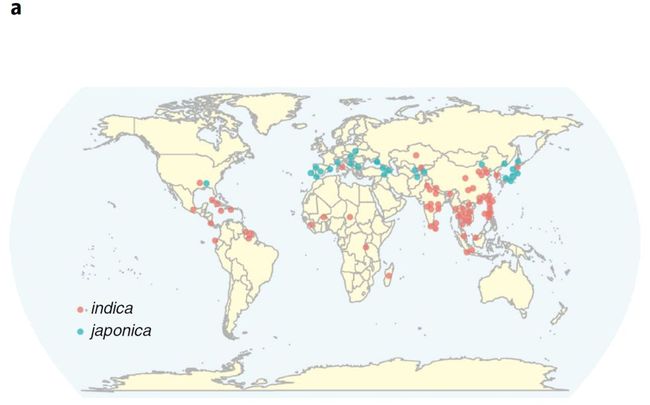
a. 籼粳稻品种来源地图(样本来源包括44个国家),红色代表indica籼稻,蓝色为japonica粳稻。
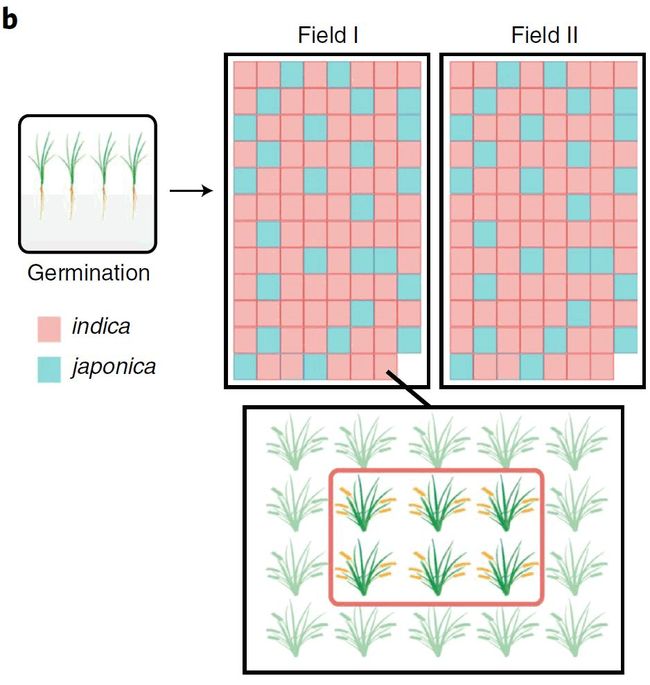
b. 水稻田间试验的实验设计图。籼粳稻品种种植位置随机排布。收获的每个水稻品种,周围作为保护行以区分不同品种。
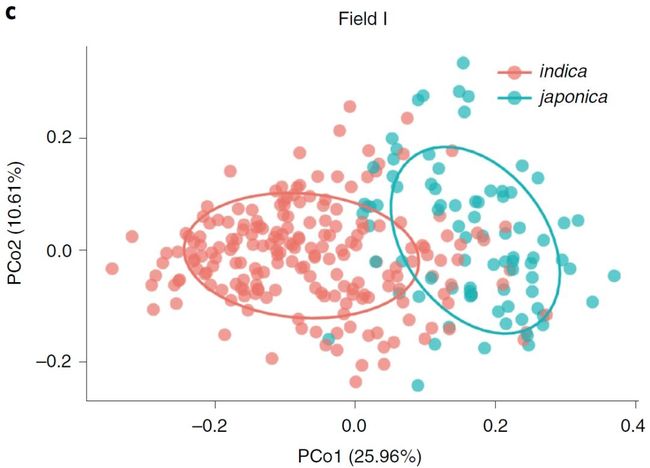
c. 基于Bray-Cutis距离的主坐标轴分析(PCoA)表明籼粳稻的根系微生物组在第一主轴分开( P < 0.001,PERMANOVA采用Adonis函数转换检验)。椭圆包括亚种68%的数据。
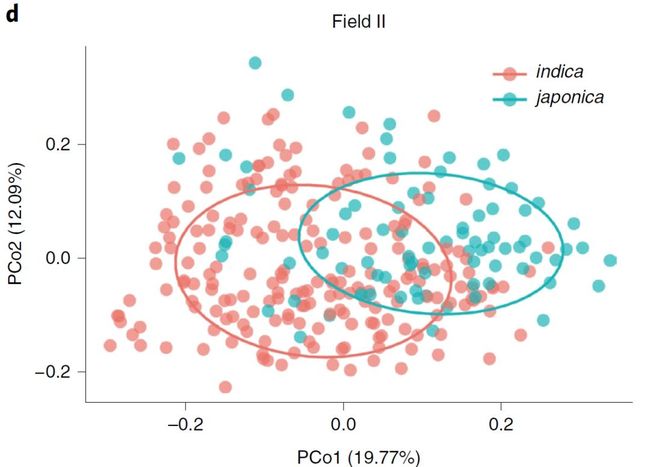
d. 基于Bray-Cutis距离的PCoA在地块2中结果表明籼粳稻根系微生物组也在第一主轴分开。
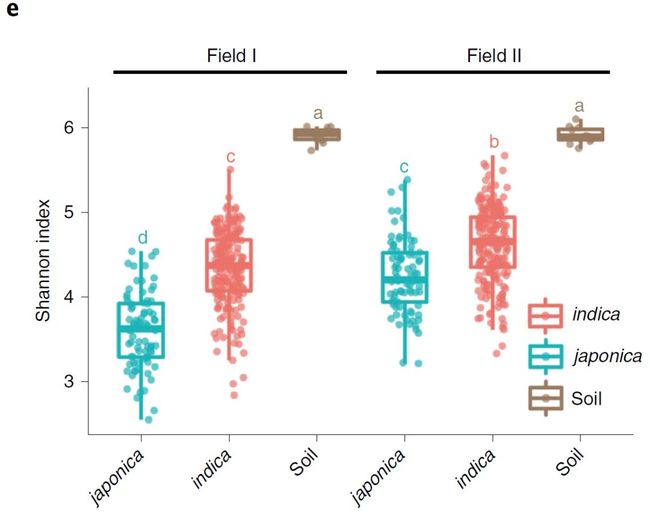
e. 箱线图展示籼粳稻和土壤的香农多样性指数。箱体上中下线分别为75、50(中位数)和25分位数。字母用于区分组间是否存在显著区别(ANOVA, Tukey-HSD)。
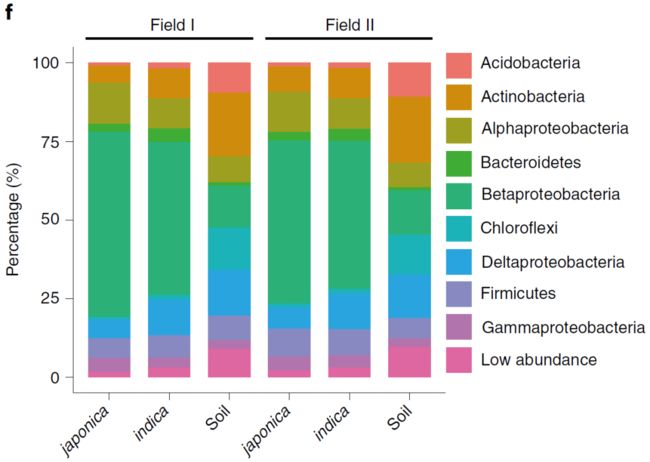
f. 展示两块地籼粳稻门水平的组成。图中的样本量如下:地块1:籼稻 (n = 201), 粳稻 (n = 80), 土壤 (n = 12); 地块2, 籼稻 (n = 201), 粳稻 (n = 81), 土壤 (n = 12)。
根系微生物组作为区分籼粳稻亚种的生物标志物
Root microbiota as biomarkers for indica and japonica
我们分析水稻的根系微生物是否如籽粒形状、叶颜色、基因组特征等一样,可以作为鉴别籼粳稻的生物标志物。我们基于地块2的根系微生物组数据,采用机器学习的随机森林方法,在门、纲、目、科、属和OTUs层面分别建立预测模型,其中在科水平判别的准确率高达83.7%(附表5)。采用10X交叉验证并重复5次评估生物标记的贡献度。交叉验证错误率在18个特征时稳定。因此我们选择了这18个科作为生物标记(图2a)。其中15个为籼稻富集,3个为粳稻富集(Wilcoxon秩和检验,采用FDR校正的P值 < 0.05,图2b)。籼稻富集的特征更多与籼稻拥有更高的微生物多样性结果一致。
我们将地块I的样品作为测试集,应用随机森林模型预测准确率为86%,其中籼稻预测准确率为94.5%,粳稻的准确率为77.5%(图2c;附表5)。为了进一步验证模型的普适性,我们在距离海南陵水农场2,482公里外北京的两个农场(昌平和上庄)种植的水稻品种IR24(籼稻)和ZH11(粳稻)进行预测,这些品种不包括在海南种植的95个品种中,结果仍然较为准确(图2d;附表5)。根系微生物被地理位置显著影响,而我们的模型适合于过去两年的六块地。未来纳入来自更多地点、更多品种的数据可能进一步改善模型的准确率。以上结果表明,根系微生物可以作为生物标记来区分籼粳稻亚种。
图2. 随机森林准确预测籼粳稻亚种
Fig. 2 | Random-forest model detects bacterial taxa that accurately predict indica and japonica subspeciation.
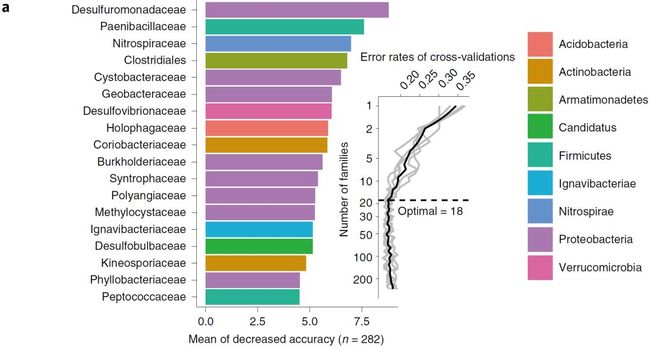
a. 基于地块2中籼粳稻科水平相对丰度建立随机森林模型中贡献度最高的18个科。生物标记分类按贡献度降序排列。插图代表10倍交叉验证错误率以评估特征贡献度和选择适合的特征数据。图例为科按门水平着色。
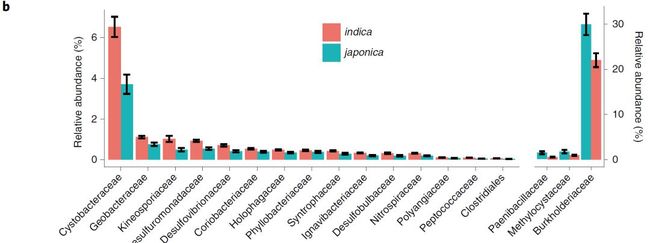
b. 重要科生物标记在籼粳中的相对丰度。高度为均值,误差棒代表标准误。
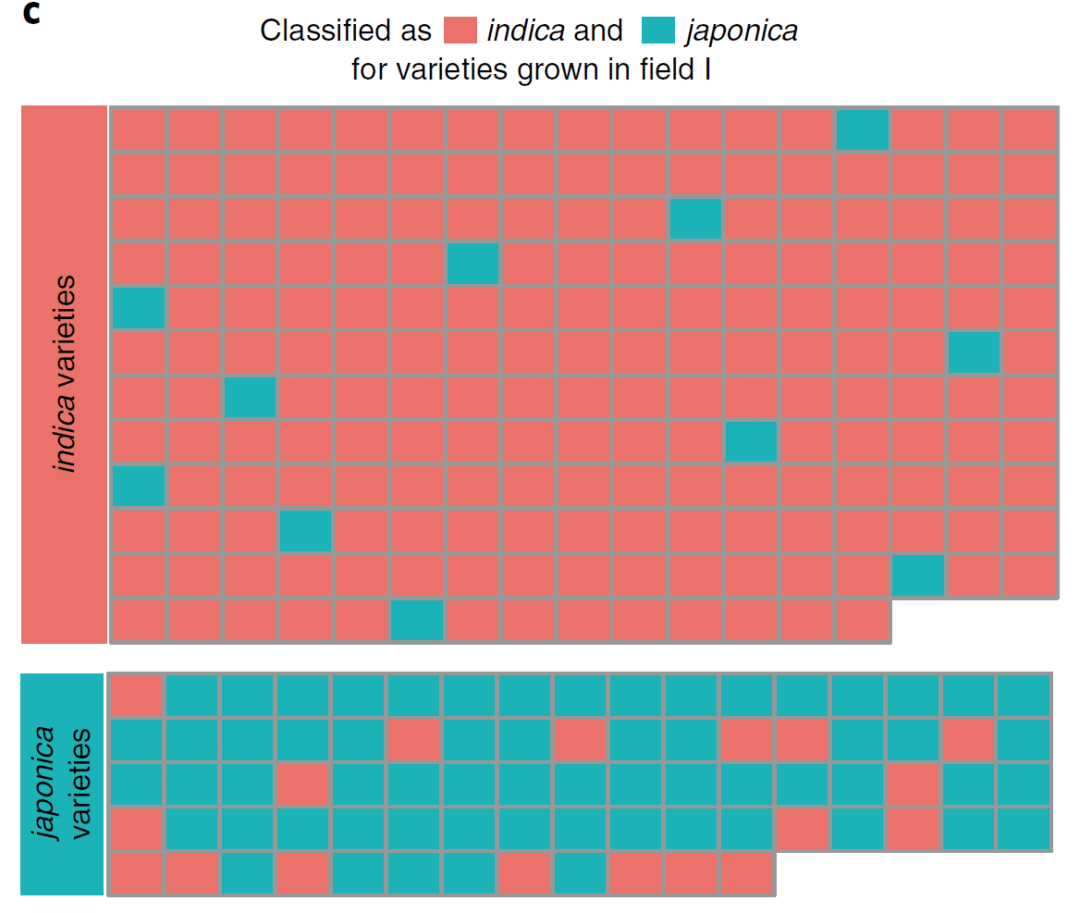
c. 对地块I中的样本进行预测的结果。每一个方块代表来自籼粳稻品种的植株。左侧为品种对应颜色,右侧为预测结果。
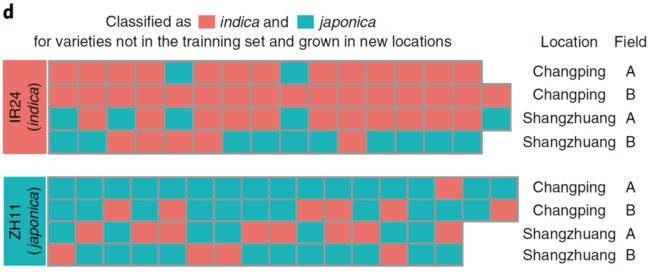
d. 对来自不同地点且非训练集品种进行预测的结果。
根系微生物组和氮利用
Root microbiota and nitrogen use
接下来我们挖掘籼粳稻在OTU层面的差异。首先,我们采用曼哈顿图查看OTUs在分类学上的分布(图3a/b;附表6)。在两块地中,籼稻富集的OTUs分布于多个门,包括产酸菌门(Acidobacteria)、变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、绿弯菌门(Chloroflexi)、厚壁菌门(Firmicutes)和疣微菌门(Verrucomicrobia)(Wilcoxon秩和检验,FDR < 0.05,图3a;附表6)。而粳稻富集的OTUs主要属于变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)和厚壁菌门(Firmicutes)(图3a,b;附表6)。其次,我们发现在两块地点籼稻富集的OTUs存在极大的比例重合(图3c,d):141个籼稻富集的OTUs(占地块I籼稻富集OTUs总量的57.8%;地块II的95.9%)在两个地间共有,其中51个OTUs与土壤相比显著富集,占籼稻富含菌丰度的76.4%(图3c;附图5;附表6)。16个OTUs(占地块I粳稻富集OTUs总量的47.1%;地块II的76.2%)在两块地均表现为粳稻富集,与土壤相比其中14个在粳稻根系富集,占粳稻富集菌丰度的99%(图3d;附图5;附表6)。籼粳稻间大多数相对丰度差异的OTUs在两个测试地点保持一致。
我们发现许多籼稻富集OTUs的功能与氮循环相关。我们采用FAPROTAX数据库(包括已发表文献中报导的细菌有关元素循环的功能,详见《功能预测之元素循环——FAPROTAX使用教程》 )注释在两块地中共有的籼粳稻富集OTUs。这些OTUs与37个通路相关(附图6)。明显的是,大多数籼稻富集的OTUs(25.5%, 36/141 OTUs)与氮循环相关,尤其在硝酸盐氨化、硝酸盐反硝化、硝酸盐还原、硝化和亚硝酸盐氨化通路中显著富集(置换检验 P = 0.025,图3e,g;附表7),反映出水稻淹水土壤中复杂的氮形式。在OTUs累计丰度角度,籼稻富集的35个氮相关菌丰度为11.7%,高于粳稻c富集的5个氮相关OTUs丰度3.4%。此外,在两块地中表现为籼稻富集的9个最高丰度的功能中,6个与氮循环相关,丰度占籼稻富集功能的53.3%(图3i,j;附表7),表明氮转化在籼稻的根系环境中比粳稻更活跃,这与之前报导的籼稻氮利用效率更高是一致的。值得注意的是,氮循环相关的OTUs在田间水稻生长后期相对丰度增长(图3f,g;附表7),表明植物水稻可能与环境微生物群体活跃合作,以调节土壤中养分来优化植物的生长。
图3. 籼粳稻根系微生物组物种和功能的差异
Fig. 3 | Taxonomic and functional characteristics of differential bacteria between the indica and japonica root microbiota.
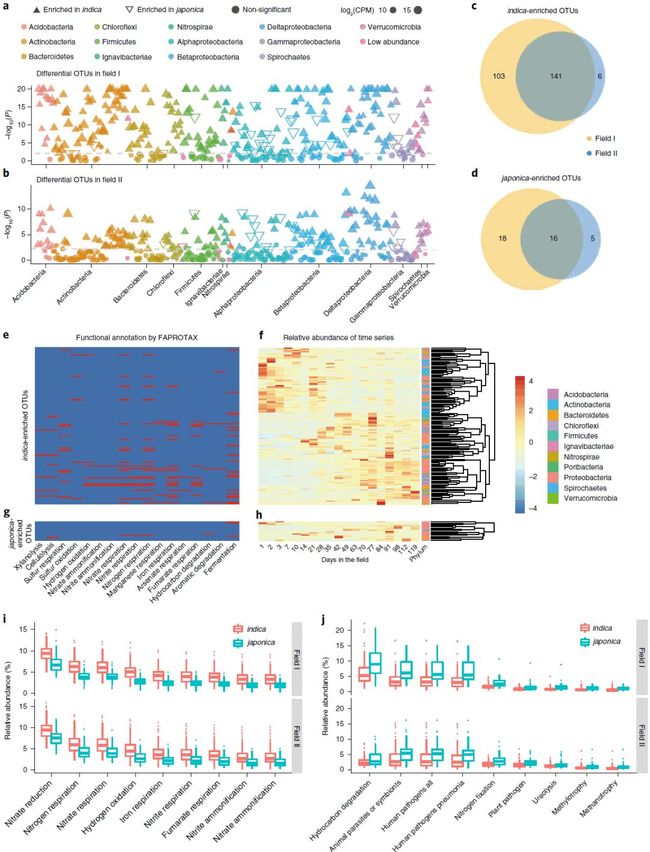
a/b. 曼哈顿图展示地块I(a)和地块II(b)籼粳稻间差异的OTUs。圆形或三角形代表OTUs,籼稻显著富集的为实心上三角,粳稻显著富集的为空心下三角,采用Wilcoxon秩和检验,阈值为FDR校正的P值 < 0.05。OTUs在图中按物种注释字母顺序排列,按门和变形菌纲着色。
c/d. 籼稻(c)和粳稻(d)在两块地共同富集的OTUs。
e/g. 基于FAPROTAX注释两块中共有籼稻(e)和粳稻(g)中特异富集的OTUs。每行代表一个OTU,红色代表有文献报导包括此类功能。
f/h. OTUs对应的北京昌平农场种植的水稻时间序列样品数据中的相对丰度,列代表取样天数。
i/j. 代表基于FAPROTAX注释功能条目中,籼稻(i)和粳稻(j)特异富集的功能通路累计相对丰度。
籼粳稻间根系微生物组差异与NRT1.1B基因
NRT1.1B and root microbiota variation
NRT1.1B基因已报导的功能是氮转运蛋白和受体,在籼粳稻差异的氮利用效率中起作用。我们假设籼粳稻根系微生物组的差异受NRT1.1B调控。研究结果表明NRT1.1B自然变异的SNP与根系微生物组差异的氮代谢功能相关(地块I中P = 2.2 X 10–16; 地块II中P = 1.8 X 10–12, 双尾t-test, 图4a; 附图7),表明NRT1.1B可能参与水稻根系微生物组的建立。
为研究NRT1.1B在水稻根系微生物组建立中的作用,我们采用16S扩增子测序北京昌平农场种植的野生型中花11和nrt1.1b缺失突变体的根系微生物。发现NRT1.1B与籼稻富集OTUs的丰度相关。基于Bray-Curtis距离的PCoA显示nrt1.1b突变体与野生在第1/2主轴分开,表明NRT1.1B在水稻根系微生物组形成中起重要作用(图4b;附图8)。在另一套完全独立的时间和地点的重复实验,结果也类似(附图8)。此外,nrt1.1b突变体中下调的OTUs与籼稻中富集的OTUs显著重合(图4c,e;附图9;附表8)。例如nrt1.1b突变体中下调OTUs的67.6%(176个中的119个)至少与一块点中籼稻富集的OTUs重合,49.6%(70个中的141个)双块地同时籼稻富集的OTUs与如nrt1.1b突变体中下调的OTUs重合,这部分占籼稻富集OTU总丰度的86.5%(附表8)。许多细菌参与氮循环,包括在籼稻中相对丰度最高的厌氧粘细菌(Anaeromyxobacter)有报导其具有氨化作用,在nrt1.1b突奕体中丰度下降(图4c,e;附表8),暗示着NRT1.1B参与招募水稻根系很大比例籼稻富集的细菌。
我们同时也对NRT1.1B在籼粳稻中不同形式的近等基因系在昌平农场进行研究(图4b;附图8)。籼稻形式的NRT1.1B特异富集7个OTUs,有6个与两块地中籼稻富集的OTUs一致(图4d,f;附图8和9;附表8)。这6个OTUs占籼稻富集OTUs总丰度的29%(附表8)。这些结果提供了NRT1.1B与水稻根系微生物组建立联系和贡献籼粳稻根系微生物组分化的进一步证据。
为进一步探索NRT1.1B在水稻微生物组建立中的功能,我们采用宏基因组测序野生型中花11和nrt1.1b缺失突变体的根系微生物。在去除宿主序列后,每个样品获得了平均30.8 Gb的微生物序列(原始序列保存于北京基因组所数据中心,编号CRA001362)。我们采用从头组装的策略拼接这些序列,并预测基因,然后采用KEGG数据库进行基因功能注释。对其中基因与氮循环通路相关的条目进行筛选和分析,与之前功能预测的结果一致,即在野生型中花11和nrt1.1b缺失突变体的根系微生物中几个氮循环相关的基因表现出相对丰度的差异(附表9)。例如,氨化过程的三个关键基因(NapA、NapB和NirD)的相对丰度在nrt1.1b突变体中下调了2.8-5.9倍,而在通路中的其它基因没有显著不同(图4g;附表9)。NapA和NapB编码硝酸还原酶,可将硝酸盐变为亚硝酸盐;NirD参与转化亚硝酸盐为铵。这些数据表明NRT1.1B与细菌根系细菌的相对丰度相关,其中包括氨化途径的关键基因:这些根系微生物可能在根系环境催化反应合成氨。
图4. NRT1.1B贡献籼粳根系微生物差异
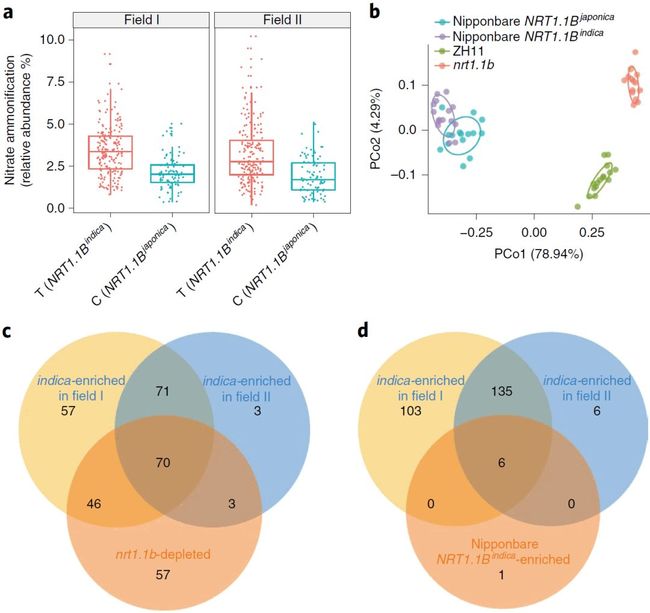
a. 群体水平 NRT1.1B 的自然变异(980位置在籼稻为T,粳稻为C,对应氨基酸为Met237Thr)与氨化功能相关(双尾t检验地块I的 P = 2.2 X 10–16 和地块二 P = 1.8 X 10–12)。
b. 基于Bray-Cutis距离PCoA显示NRT1.1B野生型与突变体,和近等基因系在第一、二主轴分开,每个点为一个样品。
c. nrt1.1b突变体中下降OTUs与两块地点籼稻富集菌重叠,三者重叠的OTUs对应的物种组成见e。
d. NRT1.1B近等基因系中籼稻形式富集的OTUs与两块地点籼稻富集菌重叠,三者重叠的OTUs对应的物种组成见f。
g. NRT1.1B野生型ZH11和突变体nrt1.1b与水稻根系宏基因组中氨化通路基因差异相关。
培养水稻根系微生物组
Cultivation of rice root microbiota
为了进一步研究籼、粳稻根系微生物组中特异富集OTUs的功能,我们基于昌平农场种植的籼稻品种IR24和粳稻品种日本晴进行细菌分离培养,以建立可用细菌研究材料资源库。为了最大可能的分离IR24和日本晴根系招募的细菌,每个品种种植于两个地点。对两地点的样本混合,采用梯度稀释的方法在96孔细胞培养板中对细菌进行分离培养(在线方法)。基于之前454平台的鉴定方法,我们进立一了种两步标签靶向V5-V7区16S核糖体基因结合Illumina高通量测序鉴定物种分类的方法(附图10)。与之前454测序方法中将一板中有标签的孔中PCR产物混合的方法相比(附图11),我们的方法对每个孔单独培养的细菌进行鉴定,可有效的避免不同板、孔间的嵌合体形成(附图10)。同时部分根也进行非培养的16S扩增子测序,用于评估培养细菌的种类和丰度比例。
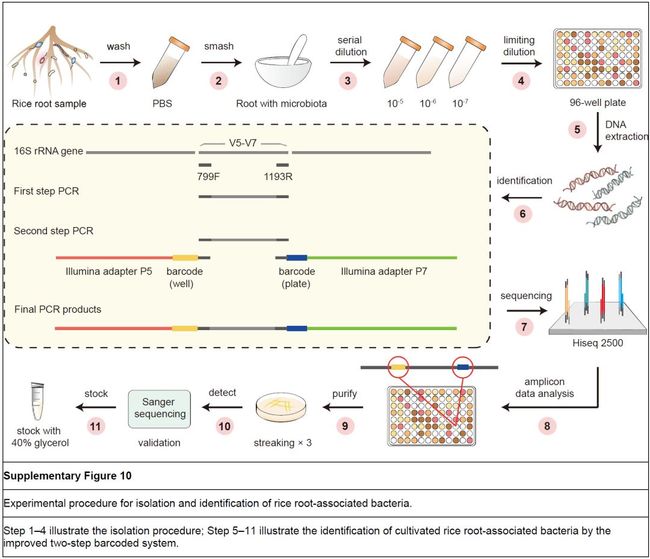
附图10. 两步PCR加双端barcode高通量细菌鉴定方法流程图
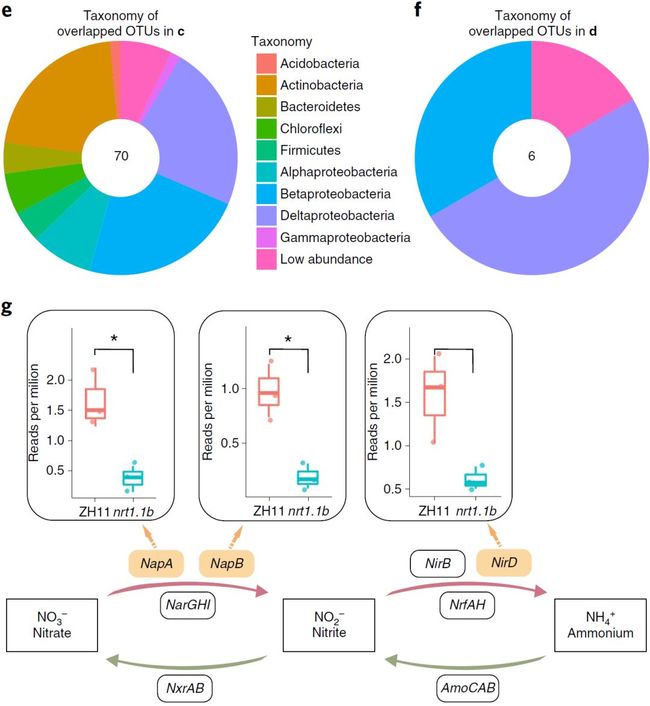
在IR24和日本晴分离培养物中总共获得了13,512菌落形成单位(CPUs,附表10)。移除低质量(Q < 20)序列,Unoise算法去除嵌合体和低丰度序列(n<8),共获得了1041条非冗余细菌16S rRNA基因序列,它们占籼稻自然样本16S鉴定种类的68.8%,和粳稻的71.7%(**图5a,b**展示至少一个样品中相对丰度 > 0.1%;附表10)。我们鉴定根系来源的CFUs包括了常见的四大菌门和27个科与水稻根系相关。因此,绝大数数与水稻相关且可重复的细菌获得了可培养的成员。
我们从13,512个CFUs中筛选有代表性的细菌核心菌株,并进行纯化和桑格测序16S rRNA基因。为了增加培养资源库的种内多样性,对16S序列相同但来源不同的菌株进行保留,因为这代表着独立的宿主起源事件(图5c)。我们总计获得1079个根系起源的隔离群(isolates),包括22个细菌目(图5c;附表11),对将来研究水稻根系细菌群落功能成为可能。
图5. 培养获得根系相关细菌种类的大多数
Fig. 5 | Rice root-associated bacterial culture collections capture
the majority of bacterial species that are reproducibly detectable
by culture-independent sequencing.
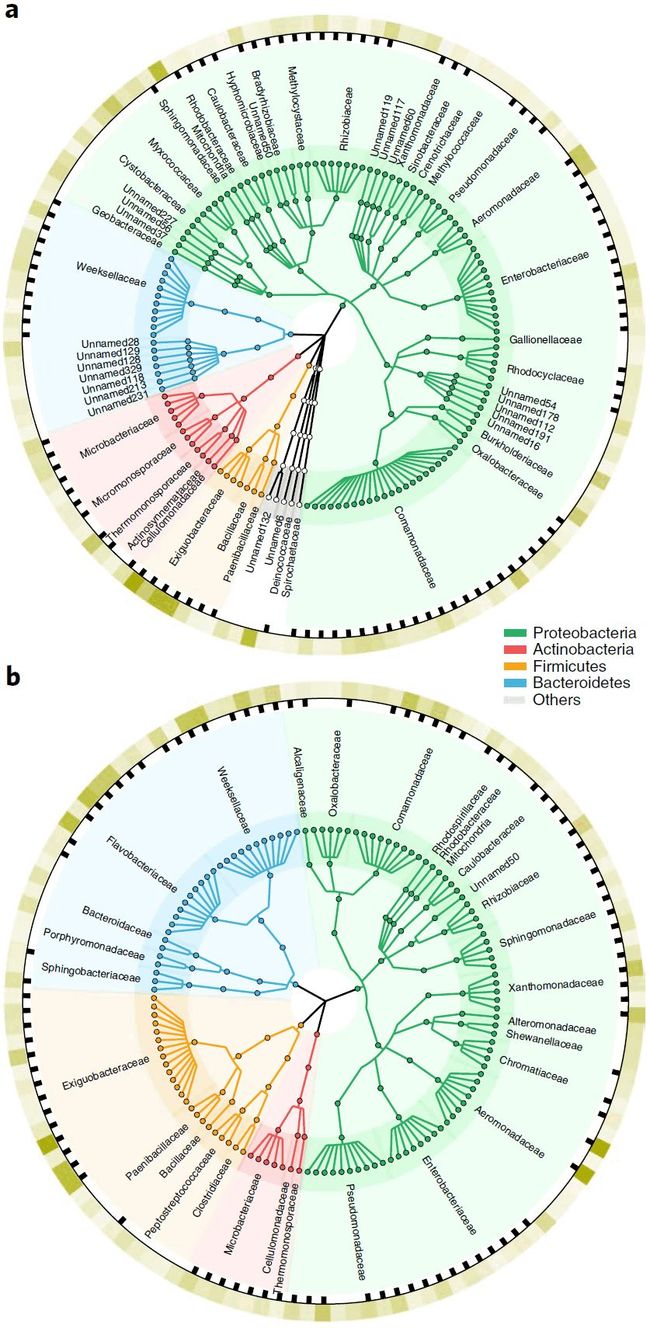
a. 以籼稻品种IR24为材料分离培养的根系细菌。内环代表根系16S非培养测序的OTUs(仅丰度大于千分之一展示在图中),外环黑块表示获得了可培养的菌,最外环为16S测序获得的相对丰度;
b. 粳稻品种A50为材料分离培养的根系细菌。
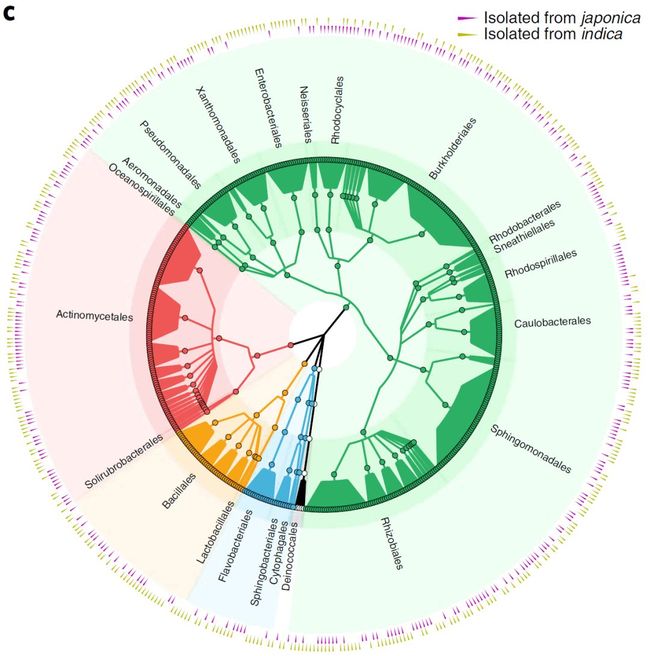
c. 对1079个纯菌中515个非冗余16S序列的物种分类层级关系图。外圈用黄色和紫色分别代表分离来源于籼稻或粳稻。
合成群落改进水稻氮吸收
Synthetic communities improve nitrogen assimilation in rice
为探究籼粳稻富集细菌的氮相关功能,我们使用灭菌煅烧土(clay)基质的无菌苗系统,添加人工合成群落(SynCom)以验证细菌群落的功能,分别添加可准确控制的硝酸盐、铵盐和可溶有机氮(附表12)。我们设计的SynCom分别包括籼、粳稻富集且与NRT1.1B相关的OTUs。选择培养的细菌与OTUs至少相似度大于97%的细菌种类用于实验。在硝或铵充足的条件下,籼、粳稻富集的SynCom抑制了水稻品种IR24的生长,可能是与水稻竞争了无机氮(附图12,13;附表13)。我们发现籼稻富集的菌在以有机氮为唯一氮源时显著促进IR24生长(图6;附表13)。与包括16个籼稻富集SynCom共培养16天,IR24的根更长(P = 1.29 X 10–12, ANOVA, Tukey-HSD)和地上部分更大(P = 5.74 X 10–12)。与此同时,包括3个粳稻富集菌的SynCom也能促生长但弱于籼稻(图6;附表13)。土壤富集的细菌,灭活的籼、粳稻富集菌没有促生效果(图6;附表13)。这些初步的研究结果表明籼稻富集细菌在有机氮的相关功能可能促进籼稻更高的氮利用效率。
图6. 籼稻富集菌在有机氮下促进水稻生长
Fig. 6 | Indica-enriched SynCom have stronger ability to promote rice
growth under the supply of organic nitrogen than japonica-enriched
SynCom.
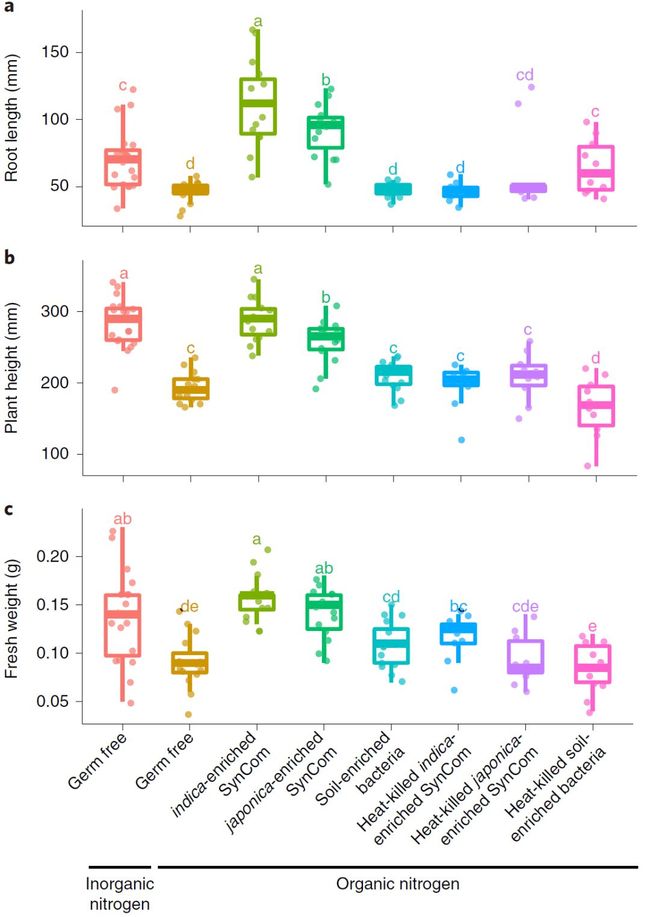
水稻品种IR24种植于无机氮和有机氮(混合5种氨基酸,附表12)条件下,并分别添加籼稻/粳稻/土壤富含菌和灭活对照。生长两周后测序水稻的根长(a)、株高(b)和鲜重(c)。
讨论
在长期的种植历史过程中,亚洲两个主要水稻栽培亚种(籼稻和粳稻)拥有不同的基因型和表型。在这里,我们在田间条件对籼稻和粳稻多个代表品种的根系微生物组的差异进行研究,发现籼粳稻品种招募明显不同的根系微生物组(图1)。我们发现一些微生物可以作为生物标记物来区分水稻亚种(图2)。此外,我们报导了NRT1.1B与水稻根系招募亚种特异的微生物相关(图4),表明在固定的环境下宿主对微生物组形成具有重要作用。NRT1.1B的自然变异可解释亚种间29%的差异丰度,因此将来鉴定其它调控根系微生物组成差异的基因至关重要。
在自然环境中,根系微生物组对宿主植物生长至关重要。我们发现籼稻品种招募较高比例氮循环相关细菌(图3),表明籼稻比粳稻根际更高的氮转化效率,很好的解析了之前籼稻更高氮利用效率的现象。基于培养的细菌和使用人工重组群体(synthetic community, SynCom)技术,发现在补充有机氮源时籼稻富集菌对水稻生长促进更加明显(图6)。我们猜测是SynCom将有机氮转化为无机氮,方便水稻根系高效吸收。更进一步的SynCom实验有助于鉴定籼、粳稻特异富集细菌的功能。
有趣的是,水稻亚种根系微生物的建立和差异与氮转动和受体基因NRT1.1B相关。因此,水稻根系可能利用转运蛋白负责氮吸收来指示土壤中硝酸盐浓度,并调控氮相关基因或根系代谢,从而影响根系微生物组的招募。进一步研究拟南芥中的NRT1.1B (chl1-9)有助于揭示该氮转运蛋白和受体在根系微生物组建立过程中的作用。
我们的数据表明植物中的水稻与根系微生物群落合作,以获取土壤环境中的氮素来优化农业条件下的生长。根系微生物、土壤氮循环、基因调控和宿主植物性状间的关系打开了技术调节根系微生物组以增加作物产量和可持续农业的道路。
方法
Methods
植物萌发、移苗和土壤成份测定
Plant germination, transplantation and measurement of soil properties
对95个栽培品种(68个籼稻和27个粳稻)的种子进行脱壳,然后再进行表面消毒:75%乙醇表面消毒30 s,2.5%次氯酸钠处理15 min共3次。然后在MS琼脂培养基上进行发芽。从44个国家收集的籼稻和粳稻品种代表了水稻的多样性(附表2)。水稻萌发15天后,在海南省陵水农场(北纬 18.512°,东经 110.044°),将秧苗移栽到两个被1.5米宽的小路间隔的水田中。这两块地过去10年来一直用于水稻的种植。近3年来,栽培方式有所不同(附表1)。第一块地种植一种高品质的杂交水稻品种——培杂1303(Peiza1303),它需要相对较低的氮肥投入(每100平方米1公斤氮)。第二块地种植一种高产杂交水稻——特优009(Teyou009),它依赖高氮肥投入(每100平方米2公斤氮肥)。
2017年1月,籼稻和粳稻亚种共包括的95个品种在氮肥投入每100平方米1.5千克氮的每块农田中随机种植。从3个土壤样品中测量了土壤的物理和化学性质,这些土壤样品在每个田地的未种植区域的 5 - 6 cm 深度处采集。采样点分布均匀。采用烘箱加热法测定有机物。土壤中有效氮、磷和钾的测定是根据前人的文献,采用二乙烯三胺五乙酸(DTPA)萃取法测定土壤微量营养素浓度。将10 g风干土壤与20 ml DTPA提取液(0.005 M DTPA,0.01 M CaCl2,0.1 M 三乙醇胺,pH 7.3)在室温下摇动2 h混合。过滤液体上清液,然后通过电感耦合等离子体发射光谱法(ICP-OES,Optima 7300 DV,Perkinelemer)分析铁、锰、铜和锌的浓度。附表1列出了土壤分析物的详细信息。
水稻栽培、样品采集和表型调查
Rice cultivation, sample collection and plant traits
每一个水稻品种都生长在20(4×5)颗水稻植株的地块上,每个植株之间的间隔为20厘米(图1b)。两品种间块地之间的距离为30厘米。地块边界上的植物是为保护行,不用于采样和收获。在分蘖后期,幼苗移栽到田间8周后采集根系样品。从一个中心位置选择每种水稻的三个代表个体。摇动根以去除松散粘附的土壤,然后冲洗,直到没有可见的土壤颗粒。将距地面10 cm长的根切成2 mm的小段,并放入2 ml试管中。本研究共使用了563份水稻根系样本。水稻品种详见附表2。在每一块土地上,从未种植的土壤收集12个相应的土体土壤样本。所有样品立即储存于-20°C,用干冰运输至实验室,储存于-80°C。2016年和2017年,在昌平农场(北纬 40.109°,东经 116.424°)种植NRT1.1B相关材料,种植方式和采样方法与上述相同,每个基因型每次收集15个独立生物学重复的野生型(ZH11)、突变体、受体亲本(Nipponbare)和近等基因系进行细菌16S rRNA基因分析。
DNA提取、PCR扩增和测序
DNA extraction, PCR amplification and sequencing
采用Illumina测序,对水稻根系及相应的土壤样品进行细菌16S rRNA基因测序。用FastDNA SPIN Kit(MP Biomedicals)提取每个样本的DNA。用PicoGreen
dsDNA Assay Kit(Life Technologies)测定DNA,然后稀释至3.5 ng μl–1。用简并引物799F和1193R扩增细菌16S rRNA基因的V5-V7区。在30 μL反应体系中,将每个样品扩增三份(水作为阴性对照),其中包括3μL稀释DNA、0.75 U PrimeSTAR HS DNA聚合酶、1个Primastar缓冲液(Takara)、0.2 mM脱氧核苷三磷酸(dNTPs)和10 pm 含条形码的正向和反向引物。在98°C的初始变性步骤30 s后,将目标区域扩增25个循环:98°C,10 s、55°C,15 s、72°C,60 s,然后在72°C下进行5分钟的终延伸。如果阴性对照组没有可见的扩增(未添加模板),则混合三份技术重复PCR产物,并使用AMPure XP Kit(Beckman Coulter)纯化。用Nanodrop(Nanodrop 2000 C,Thermo Scientific)测量纯化的PCR产物,并稀释至10 ng μl–1作为第二轮PCR的模板。所有样品用第二轮引物(附表14)扩增三次技术重复,条件与PCR第一次相同,但仅有八个循环。将每个样品的技术重复合并,并在浓度1.2%(W/V)的琼脂糖凝胶上电泳,细菌16S rRNA基因扩增子使用QiaQuik凝胶提取试剂盒(QiAGEN)进行胶回收。随后用PicoGreen dsDNA分析试剂盒(Life Technologies)测量DNA,并将每个样品的200 ng混合。最终的扩增子文库使用Agencourt AMPure XP试剂盒(Beckman Coulter)纯化两次,并在Hiseq 2500平台(Illumina Inc.)的双端250 bp(PE250)模式上进行测序。
16S rRNA基因扩增子分析
Bioinformatics analysis on 16S rRNA gene profiling
使用QIIME v.1.9.1、usearch v.10.0和自编写脚本处理16S rRNA基因序列(详见《扩增子分析流程-把握分析细节》)。样本元数据见附表3。使用fastqc v.0.11.5对双端序列的质量进行评估,并在以下步骤中使用usearch进行处理:双端序列的合并和序列按样本来源重命名(-fastq_mergepairs);剪切条形码和引物(-fastx_truncate);过滤低质量序列(-fastq_filter);以及获得非冗余序列(-fastx_uniques)。非冗余序列按97%相似性聚类为OTU。代表性序列由uparse选择。将OTU与Silva132数据库比对,去除嵌合体和宿主质体序列。OTU表是由usearch(-otutab)生成的。用RDP分类器对代表性序列进行分类。利用自编脚本和QIIME2进行多样性分析。利用Wilcoxon秩和检验对每种基因型相对丰度中位数大于0.2%的OTU进行丰度和分类差异分析,FDR对多重检验的相应P值进行校正,筛选显著性的阈值为FDR < 0.05。利用FAPROTAX v.1.1(《元素循环FAPROTAX教程》)对原核类群进行功能注释。水稻时间序列数据来自我之前发表的论文(《手把手带你重现菌群封面文章图表——水稻时间序列分析》)。为了获得籼稻和粳稻品种分类群的最佳鉴别性能,我们在R语言中使用randomForest package v.4.6–14默认参数对水稻亚种的门、纲、目、科、属和OTU水平的细菌分类群的相对丰度进行了分类,以确实最优的分类层级,发现科水平效果最好。以地块二数据为训练集,利用randomForest (importance = TRUE,
proximity = TRUE)函数生成籼粳分类模型。通过rfcv()函数执行交叉验证,以选择适当的特性。用varImpPlot函数导出特征在分类中的重要性。使用R v.3.5.1中的ggplot2 v.2.2.1包可视化特征的重要性和交叉验证曲线。在默认参数下,使用地块一和北京栽培的部分品种的数据作为模型的验证集。
宏基因组分析
Metagenome analysis
使用双端100碱基(PE100)测序策略,在BGISEQ-500平台上对3个ZH11根样本和3个nrt1.1b突变样本的DNA进行测序,每个样本的平均值为44 GB。所有原始数据均由Soapnuke v.1.5.2进行质量控制,然后根据Oryza sativa基因组(GI:996703420;登录号:NW_015379174.1),使用使用Soap2.22对干净序列进行去除宿主污染,过滤后每个样本平均30.8 GB。由Megahit v.1.0进行组装,在接下来的分析中丢弃长度小于200 bp的组装重叠群(contig)。过滤后的重叠群用blastn-2.2.31与Oryza sativa基因组进行比对,从重叠群层面二次去除宿主污染。基因注释通过使用MetaGeneMark_v1_mod预测,保留长度大于或等于100 bp的基因。所有预测基因合并在一起,然后使用CD-HIT聚集成一个非冗余的参考基因集,设置相似性阈值为95%。通过BLASTX-2.2.31对KEGG v81进行非冗余基因注释,并保留符合标准相似度>30%和E值评分<1×10-5的结果。接下来,使用soap2.22将干净的序列与非冗余基因集比对,并根据比对序列的数量计算基因的相对丰度。通过对同一个基因的丰富性进行总结,生成了KEGG直系同源簇(KO)组成。采用STAMP软件中的Welch’s t检验和FDR校正KO相对丰度的差异的统计为P值。水稻单核苷酸多态性数据是从先前Molecular Plant发表的论文中下载的。用t检验评价NRT1.1b中的功能单核苷酸多态性与氮相关功能之间的相关性,并在R中的ggplot2进行可视化。
根源性细菌的分离
Isolation of root-derived bacteria
通过梯度稀释和基于16S rRNA基因的群体分析,在不同的自然土壤中种植水稻进行细菌分离。为了获得具有代表性的根定植细菌库,我们使用了两个具有代表性的水稻品种IR24(籼稻)和Nipponbare(粳稻)。每个品种在昌平农场(中国北京,东经116.424°E,北纬40.109°N)的两个田间种植,植物在开花前就收获。将同一品种的三棵健康植株的根系在各田间混合,以减少其生物学变异。在180 转/分钟的振动器上去除土壤颗粒,在洗涤缓冲液(PBS)中漂洗水稻根三次,每次持续15分钟,然后将每个根中在匀浆机中打碎15分钟,取上清液稀释于1:10(v/v)胰蛋白酶大豆肉汤中,并滴于96孔的微量滴定板中,室温下培养20 天。在分离根源细菌的同时,对生长在上述土壤中的水稻植株的根进行了采集,并通过与培养无关的16S rRNA基因测序来评估细菌的多样性。
我们采用两步条形码标记引物的聚合酶链式反应(PCR)技术扩增,结合Illumina Hiseq测序确定了根源细菌16S rRNA基因的V5-V7序列。在含25 mM NaOH和0.2 mM EDTA的10 μL缓冲液I中,加入6 μL细菌培养物溶解于缓冲液,在95°C下的pH 12中提取30分钟,然后在pH 7.5下加入含40 mM Tris HCl的10 μL缓冲液II以降低pH 值至7。采用含有孔和板特异性条形码的简并引物799F和1193R对96孔微量滴定板中的分离株进行两步PCR,以扩增可变区V5-V7。在PCR扩增的第一步,用0.75 U-HS-Taq DNA聚合酶、10×缓冲液、0.2 mM-dNTPs(Takara)、0.1 μM正向引物(799F)和0.1 μM反向引物(1193R)(Life Technologies)在30 μL反应中扩增3 μL裂解细胞的DNA。在以下条件下进行PCR扩增:DNA在94°C下初始变性2分钟,随后30个扩增循环,每个循环在94°C下变性30秒,在55°C下变性30秒,在72°C下扩增1分钟,最后在72°C下终延伸5分钟。在96孔的PCR板中产物稀释40倍用于第二步PCR。在第二步中,用0.75 U HS-Taq DNA聚合酶、10×缓冲液、0.2 mM dNTPs(Takara)、0.1 μm 一种含有96个条形码正向引物中的一个(附表14)和0.1μm反向条形码引物在30 μl反应中扩增来自第一步的3 μl稀释PCR产物。PCR循环条件如下:首先在94°C下变性2分钟,随后25次循环94°C 30秒、55°C 30秒、72°C 1分钟,最后伸长72℃5分钟。PCR产物用Aencourt AMPure XP试剂盒(Beckman Coulter)和Wizard SV凝胶和PCR纯化系统(PROMEGA)。DNA浓度由PicoGreen dsDNA分析试剂盒(Life Technologies)测定,并以等量混合样本(附表14)。最后的PCR产物构建的文库在Hiseq 2500平台上进行测序。每个序列包含一个板条形码、一个孔条形码和v5-v7序列。根据孔和板标识符对序列进行质量过滤和样本拆分。通过UNOISE算法,去除嵌合体和低丰度序列(读取计数<8)后定义OTU(去噪方式,相当于100%相似度)。根据RDP训练集16,采用sintax算法对OTU进行分类。将培养的细菌与相应根微生物群成员中的OTU进行比较,这些根微生物群成员的16S rRNA基因相似度阈值要求大于97%则认为可培养。通过GraPhlAn 0.9.7可视化枝状物种树图展示细菌可培养的数量和种类。
水稻根系相关细菌培养库的制备
Preparation of rice root-associated bacterial culture collections
在使用单个菌落接种液体培养物之前,通过在各自的固化培养基上连续三个平板纯化,从培养细菌中鉴定出的每个非冗余OTU的代表性菌株。这些液体培养物被用于用27F和1492R引物进行桑格测序的验证,以及用于为培养物库制备甘油保藏。
植物生长促进试验
Plant growth promotion assay
水稻植株生长在含有硝酸盐、铵或可溶有机氮的粘土无菌体系中,有人工重组细菌群落或无菌对照。煅烧粘土是一种惰性的土壤替代品,用水冲洗,高压灭菌三次,热孵育至完全脱水。用75%乙醇对水稻种子进行表面消毒30秒,然后在2.5%次氯酸钠中三次消毒15分钟,并在MS琼脂培养基上萌发5天。将培养库的根源细菌在28°C的TSB培养基中50毫升试管中培养5天,然后等浓度合并,用于制作人工重组群落的接种物,但要低于根的细菌承载力。为了将重组菌群(SynCom)接种到煅烧粘土基质中,将重组菌群的光学密度OD 600调整为0.5,并将2.0 ml向心粒添加到250 ml 1 × Kimura B营养液(pH 5.8)中,并与200 g煅烧粘土在组培盒中混合(每g煅烧粘土约106个细胞)。然后将5天大的无菌水稻幼苗转移到组培盒中的粘土中。植物在25°C、16 h光照和21%湿度下生长。2周后,测定植株高度、鲜重和根长,评价植株生长促进作用。
报告摘要
Reporting Summary
有关研究设计的更多信息,请参阅本文链接的《自然研究报告摘要》。
数据可用
本文报道的原始序列数据(项目编号 :PRJCA001214)已存放在中国科学院大数据中心的基因组序列档案中,登记号为CRA001372(细菌16S rRNA基因测序数据),CRA001362(宏基因组测序数据),可在 http://bigd.big.ac.cn/gsa 上开放获取。所有细菌纯培养物(附表11)均存放在国家基因库(China
Natural Gene Bank)和中国农科院菌种保存中心(Agricultural Culture Collection of China)两个国家级菌种保藏中心。关于这些菌株的所有信息,如16S rRNA基因序列、分类和分离细节,以及任何进一步的更新,都可以在 http://bailab.genetics.ac.cn/culture_collection/ 网站上找到
代码可用
Code availability
计算分析中使用的脚本保存于Github中,网址 https://github.com/microbiota/Zhang2019NBT
Reference
Jingying Zhang, Yong-Xin Liu, Na Zhang, Bin Hu, Tao Jin, Haoran Xu, Yuan Qin, Pengxu Yan, Xiaoning Zhang, Xiaoxuan Guo, Jing Hui, Shouyun Cao, Xin Wang, Chao Wang, Hui Wang, Baoyuan Qu, Guangyi Fan, Lixing Yuan, Ruben Garrido-Oter, Chengcai Chu & Yang Bai. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nature Biotechnology. 2019, 37: 676-684. doi:10.1038/s41587-019-0104-4
https://doi.org/10.1038/s41587-019-0104-4.
王孝林, 王二涛 (2019). 根际微生物促进水稻氮利用的机制. 植物学报 54, 1–3.
Hu B, Wang W, Ou S, Tang J, Li H, Che R, Zhang Z, Chai X, Wang H, Wang Y, Liang C, Liu L, Piao Z, Deng Q, Deng K, Xu C, Liang Y, Zhang L, Li L, Chu C (2015) Variation in NRT1.1B contributes to nitrate-use divergence between rice subspecies. Nature Genetics. 47(7): 834-838. doi: https://doi.org/10.1038/ng.3337 解读 http://blog.sciencenet.cn/blog-2351-899496.html
作者简介
白洋

白洋,博士,研究员,博士生导师
2005年毕业于武汉大学,2007年获得武汉大学植物发育生物学硕士学位,2010年在德国科隆大学获得植物发育生物学博士学位,2011—2015年博士后期间在德国马克斯普朗克植物育种研究所进行植物根系微生物组学的研究。2016年至今,任中科院遗传与发育生物学所研究员,也是中国科学院-英国约翰英纳斯中心植物和微生物科学联合研究中心(CEPAMS)首位研究员目前主要研究根系微生物组在植物抗病抗逆、营养高效等过程中的功能。在Nature、Science、Nature Biotechnology、Nature Methods、Nature Plants、Microbiome、GigaScience、Science China Life Sciences等杂志发表论文20余篇。
点击以下链接,观看视频版白洋组简介
https://v.qq.com/x/page/x0842fhrc5c.html
CEPAMS官页白洋组简介视频
实验室主页:http://bailab.genetics.ac.cn/
储成才

储成才,研究员
储成才,中国科学院遗传与发育生物学研究所研究员,中国科学院大学教授,博士生导师。1999年入选中国科学院“百人计划”;2004年入选首批新世纪百千万人才工程国家级人选;2006年经国务院批准享受政府特殊津贴专家;2008年获国家杰出青年基金;2016年入选中国科学院“特聘 核心骨干”研究员,第七届全国优秀科技工作者,其领导的研究团队入选科技部国家创新人才推进计划重点领域创新团队,并荣获“第六届中国侨界贡献奖”;2017年入选中组部、人社部“万人计划”领军人才。中国科学院遗传发育所浙江嘉兴农作物高新技术育种中心副主任。
《遗传》杂志副主编,《中华科学技术大词典》、《Rice》、《植物学报》、《植物生理学报》、《农业生物技术学报》等编委。
回国后在Nature Genetics、Nature Biotechnology、Nature Plants、Nature Communications、Trends in Plant Science、Genome Research、PNAS、Plant Cell、Molecular Plant、Plant Physiology、Plant Journal等刊物发表论文120多篇(他引8158、H指数50),申请专利50多项,其中国际专利8项,与育种单位合作培育水稻新品种5个。
张婧赢

张婧赢,吉林大学本硕,中国科学院生态环境研究中心博士,遗传发育所白洋组博士后。主要研究方向为水稻微生物组。
刘永鑫

刘永鑫,博士。2008年毕业于东北农大微生物学专业。2014年中科院遗传发育所获生物信息学博士学位,2016年博士后出站留所工作,任宏基因组学实验室工程师,目前主要研究方向为宏基因组数据分析和植物微生物组。QIIME 2项目参与人,目前在Science、Nature Biotechnology、Genomics Proteomics Bioinformatics、Science China Life Sciences等杂志发表论文十余篇。2017年7月创办“宏基因组”公众号,目前分享宏基因组、扩增子原创文章800余篇,代表博文有《扩增子图表解读、分析流程和统计绘图三部曲》,关注人数4.6万+,累计阅读700万+。
张娜

张娜,河北大学生物科学本科;遗传发育所直博生。研究方向为水稻微生物组,以共同第一作者文章发表于自然生物技术、中国科学生命科学。
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读