二代测序数据统计分析中为什么是负二项分布?
本文转载自“universebiologygirl”,已获授权
1. 转录组数据统计推断的难题
在RNA-seq中进行两组间的差异分析是最正常不过的了
我们在其它实验中同样会遇到类似的分析,通常,我们可以用方差分析判定两组“分布”数据间是否存在显著差异。原理是:当组间方差大于组内方差(误差效应),并且统计学显著时,则认为组间处理是可以引起差异的。
那这不就是咱们学过的统计学里普普通通的假设检验问题吗?用熟悉的算法简单地进行计算,分分钟就能搞定吧——凸样凸拿衣服,骚年要是事情都那么简单,要科学家干嘛,问题就在于:常规方法搞不定啊!
其实统计学家也很无奈啊,看看我们转录组实验得到的这些数据吧:我们的实验只进行少得可怜的生物学重复(n<10),而且,任何基因的表达量都不能是负数,这些数据并不符合正态分布,用于表征表达量的counts是非连续的(芯片信号是连续的),RNA-seq数据的离散通常是高度扭曲的,方差往往会大于均值……,就这些奇怪的特征,使得准确估计方差并没有想象的那么容易。
我们面临两个核心问题:
基因表达数据适合用什么统计学分布进行差异显著性检验?
如何利用少量生物学重复数据估算基因表达的标准差?
2. 泊松分布 or 负二项分布?
从统计学的角度出发,进行差异分析肯定会需要假设检验,通常对于分布已知的数据,运用参数检验结果的假阳性率会更低。转录组数据中,raw count值符合什么样的分布呢?
count值本质是reads的数目,是一个非零整数,而且是离散的,其分布肯定也是离散型分布。对于转录组数据,学术界常用的分布包括泊松分布 (poisson)和负二项分布 (negative binomial)两种。
2.1. 为什么泊松分布不行?
首先有必要简单地介绍一下泊松分布
泊松分布适合于描述单位时间(或空间)内随机事件发生的次数(事件发生的次数只能是离散的整数)。如某一服务设施在一定时间内到达的人数,电话交换机接到呼叫的次数,汽车站台的候客人数,机器出现的故障数,自然灾害发生的次数,一块产品上的缺陷数,显微镜下单位分区内的细菌分布数等等。
泊松分布大概长这样:
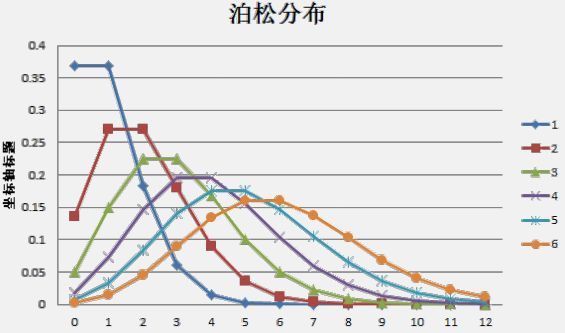
λ是波松分布所依赖的唯一参数。 λ值愈小分布愈偏倚, 随着λ的增大 , 分布趋于对称。 当λ=20时分布接近于正态分布;当λ=50时, 可以认为波松分布呈正态分布。
在数据分析的早期,确实有学者采用泊松分布进行差异分析,但是发展到现在,几乎全部都是基于负二项分布了,究竟是什么因素导致了这种现象呢?为了解释这个问题,我们必须提到一个概念 over dispersion。
dispersion指的是离散程度,研究一个数据分布的离散程度,我们常用方差这个指标。对于泊松分布而言,其均值和方差是相等的,但是我们的数据确不符合这样的规律。通过计算所有基因的均值和方差,可以绘制如下的图片:
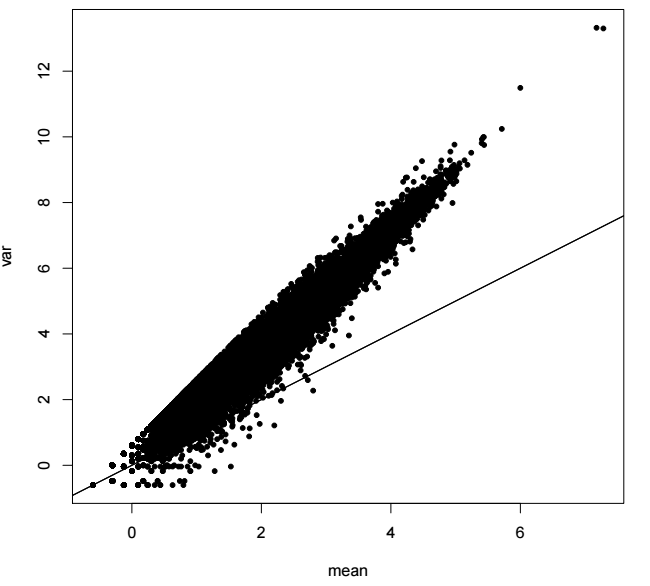
横坐标为基因在所有样本中的均值,纵坐标为基因在所有样本中的方差,直线的斜率为1,代表泊松分布的均值和方差的分布。可以看到,真实数据的分布是偏离了泊松分布的,方差明显比均值要大。
如果假定总体分布为泊松分布, 根据我们的定量数据是无法估计出一个合理的参数,能够符合上图中所示分布的,这样的现象就称之为overdispersion。
由于真实数据与泊松分布之间的overdispersion,选择泊松分布分布作为总体的分布是不合理。
以上只证明了泊松分布是个不太恰当的分布估计,那怎么证明负二项分布就是合适的分布估计呢?
2.2. 为什么负二项分布行?
主要是从均值与方差之间的关系去证明
同样的,也先简单介绍一下负二项分布:
二项分布描述的是n重伯努利实验,在n重贝努利试验中,事件A恰好发生x(0≤x≤n)次的概率为:
它的概率分布图如下:
负二项分布描述的也是伯努利实验,不过它的目标事件变成了:对于Bernoulli过程,我们设定,当某个结果出现固定次数的时候,整个过程的数量,比如我们生产某个零件,假设每个零件的合格与否都是相互独立的,且分布相同,那么当我们生产出了五个不合格零件时,一共生产了多少合格的零件,这个数量就是一个负二项分布,公式如下:
该公式描述的是,在合格率为p的一堆产品中,进行连续有放回的抽样,当抽到r个次品时,停止抽样,此时抽到的正品正好为k个的概率
它的概率分布如下:
p=0.5, r=5
负二项分布的均值和方差分别为:
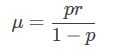
![]()
将p用μ表示,得到:
![]()
将上一步推出的p和1-p带入到方差的表达式中,得到:
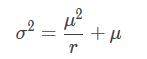
记1/r=α,则
![]()
从上面的式子可以看出,均值是方差的二次函数,方差随着均值的增加而进行二次函数形式的递增,正好符合上文 2.1. 为什么泊松分布不行? 部分均值与方差分布图的情况
其中α和r被称为dispersion parameter
负二项分布与泊松分布的关系,可以用α或r推出:
当
r -∞时,α -0,此时 σ2= μ,为泊松分布;当
r -> 0时,α -> ∞,此时overdispersion
3. 方差估计
在生物学重复很少时,我们是很难准确计算每个基因表达的标准差的(相当于这个数据集的离散程度)。我们很可能会低估数据的离散程度。
被逼无奈的科学家提出了一个假设:表达丰度相似的基因,在总体上标准差应该也是相似的。我们把不同生物学重复中表达丰度相同的基因的总标准差取个平均值,低于这个值的都用这个值,高于这个值的就用算出来的值。
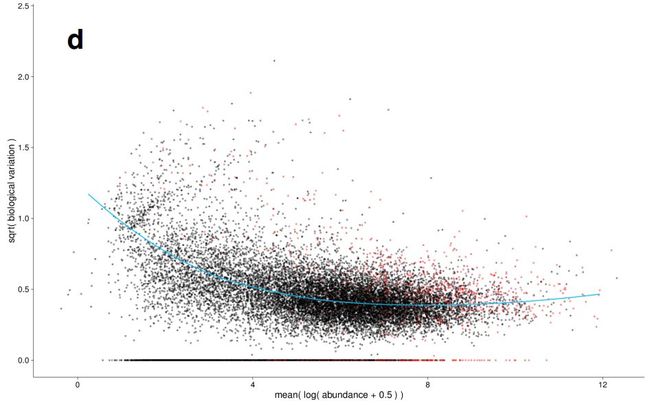
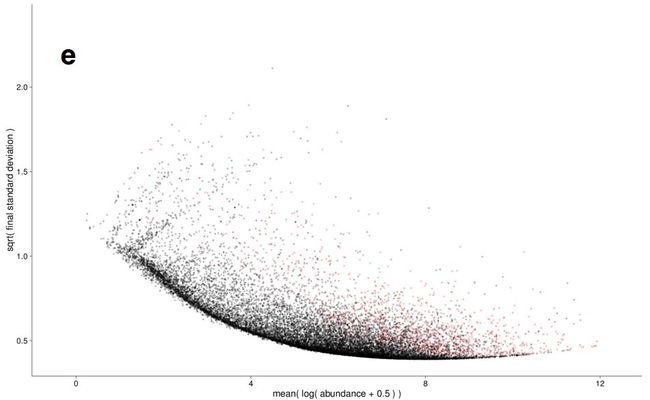 (以上图来自 H. J. Pimentel, et al. Differential analysis of RNA-Seq incorporatingquantification uncertainty. bioRxiv, 2016)
(以上图来自 H. J. Pimentel, et al. Differential analysis of RNA-Seq incorporatingquantification uncertainty. bioRxiv, 2016)
参考资料:
(1) 【生信修炼手册】负二项分布在差异分析中的应用
(2) 【 生信百科】转录组差异表达筛选的真相
(3) 【生信媛】RNA-seq分析中的dispersion,你知道吗?
(4) H. J. Pimentel, et al. Differential analysis of RNA-Seq incorporatingquantification uncertainty. bioRxiv, 2016
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
