整合QC质控结果的利器——MultiQC
一、MultiQC介绍
NGS技术的进步催生了新的实验设计、分析类型和极高通量测序数据的生成。对于这些数据的质量评估,每一步分析结果的评估是后续结果可信度的衡量和保障。不少生信工具都可以给样品生成一个评估结果,如FastQC、Qualimap 和RSeQC等 (39个转录组分析工具,120种组合评估)。但是这时又出现了一个难题,那就是几乎所有的质控工具都是针对单个样本生成一个报告,这就要求用户自己去逐一查找各个QC结果,这无疑是个十分耗时、重复又复杂的事,而且还不能快速看出所有样本的异同。
那能否把所有质控结果整合在一起呢?可以自己写程序造轮子(我们之前就是这么做的)。但现在有了MultiQC,基于Python的小工具很好地解决了这个繁琐的事情,其强大的功能主要体现在以下三个方面:
1)能将测序数据的多个QC结果整合成一个HTLM网页交互式报告,同时也能导出pdf文件;
2)支持多种分析类型的质控结果查看,如:RNAseq、Whole-Genome Seq、Bisulfite Seq、Hi-C和MultiQC_NGI;
3)支持整合68种软件分析的结果,而且支持的软件还在持续增加,也可以自己写作一个插件,具体见下图。

二、安装MultiQC
依赖python2.7+, 3.4+ 或者 3.5+
# pip安装
pip install git+https://github.com/ewels/MultiQC.git #Installation with pip
# conda安装
conda install -c bioconda multiqc # Installing with conda三、运行MultiQC
直接指定MultiQC要分析的文件路径即可,若数据在当前目录下输入multiqc .即可。
multiqc .
multiqc data/
multiqc data/ ../proj_one/analysis/ /tmp/results
multiqc data/*_fastqc.zip
multiqc data/sample_1*使用--ignore忽略掉某些文件
multiqc . --ignore *_R2*
multiqc . --ignore run_two/
multiqc . --ignore */run_three/*/fastqc/*_R2.zip四、MultiQC报告解读(以RNA-Seq数据为例)
1. General Statistics
每一个样本reads数量、比对层面的质量评估整合统计表,点击Configure Columns可以选择显示或不显示某些项。点击Plot可以绘图。

点击Configure Columns选择展示哪些项

点击Plot可以对任意两种属性的评估结果做交互式二维图,若各样本均一性好,散点会比较集中,反之会出现某些离散的点,这样方便查看某些指标异常的离群样本。

2. featureCounts
利用featureCounts工具计算每个基因外显子的reads数的结果展示。featureCounts不仅可以支持gene的定量,也支持exon, gene bodies, genomic bins, chromsomal locations的定量。功能类似的软件是HTSeq。
软件官网:http://bioinf.wehi.edu.au/featureCounts/

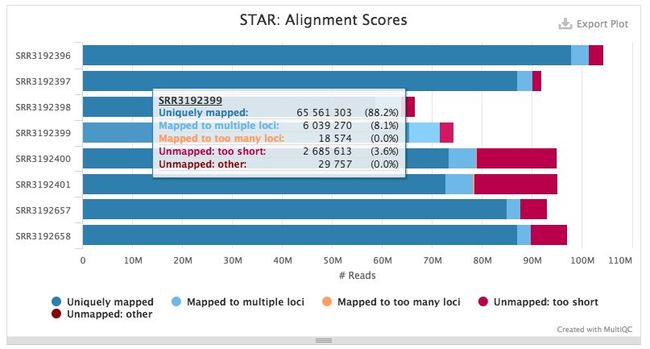
3. STAR
基于STAR比对工具的分析结果,STAR会将没有paired mapping的reads都剔除,避免single reads比对到基因组上;并且STAR对lower-quality(采用more soft-clipped和错配碱基)比对有较高的容忍度。
软件官网:https://github.com/alexdobin/STAR
更多分析工具比较见:转录组分析工具大比拼
4. Cutadapt
用cutadapt软件来对双端测序数据进行预处理,去除接头和低质量碱基。
对测序数据进行过滤时cutadapt对测序数据进行识别、剪切并去除adapters, primers , poly_A等序列,移除被adapter污染的reads部分(指由于插入片段长度不够,测序仪读到的测序引物等序列)。具体见NGS基础 - 高通量测序原理。
软件官网:https://cutadapt.readthedocs.io/en/stable/

5. FastQC
MultiQC将fastqc工具分析得到的10个结果分别整合成一个模块,集中查看。
软件官网:http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
具体的关于FastQC报告解读可以见历史推文:NGS基础 - FASTQ格式解释和质量评估

MultiQC的可定制性也比较强,更多功能值得进一步探索。
转录组研究
39个转录组分析工具,120种组合评估(转录组分析工具哪家强-导读版)
39个转录组分析工具,120种组合评估(转录组分析工具大比拼 (完整翻译版))
无参转录组分析工具评估和流程展示
120分的转录组试题(第一份答案)
120分的转录组试题(第二份答案)
120分的转录组试题(第三份答案)
DESeq2差异基因分析和批次效应移除
NGS基础和软件应用
NGS基础 - FASTQ格式解释和质量评估
NGS基础 - 高通量测序原理
NGS基础 - 参考基因组和基因注释文件
NGS基础 - GTF/GFF文件格式解读和转换
NGS基础 - 测序原始数据下载
本地安装UCSC基因组浏览器
测序数据可视化 (一)
IGV基因组浏览器可视化高通量测序数据
高通量数据分析必备-基因组浏览器使用介绍 - 1
高通量数据分析必备-基因组浏览器使用介绍 - 2
高通量数据分析必备-基因组浏览器使用介绍 - 3
测序文章数据上传找哪里
GO、GSEA富集分析一网打进
GSEA富集分析 - 界面操作
Bedtools使用简介
OrthoMCL鉴定物种同源基因 (安装+使用)
Rfam 12.0+本地使用 (最新版教程)
轻松绘制各种Venn图
ETE构建、绘制进化树
psRobot:植物小RNA分析系统
生信软件系列 - NCBI使用
去东方,最好用的在线GO富集分析工具
2018 升级版Motif数据库Jaspar
一文教会你查找基因的启动子、UTR、TSS等区域以及预测转录因子结合位点
拿到基因两眼一抹黑?没关系,先做个基因富集分析吧!
科研小萌新,掌握这些技巧,轻松玩转各个基因!
引起相变的无序结构域(IDRs)怎么预测?跟踪热点,提升文章档次!
如果你经常用PubMed,那么这个插件将非常好用!
没钱买KEGG怎么办?REACTOME开源通路更强大
美女教授带你从统计学视角看转录组分析
我想做信号通路分析,但我就是不想学编程
深度好文—单碱基编辑技术之工具篇
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
