Cell子刊:16s分析之FishTaco分析
一次偶然的机会我看到了这篇文章,发表于Cell Host & Microbe上,这个期刊也不怎么熟悉,偏机理,就点了WOS上的期刊信息:

Systematic Characterization and Analysis of the Taxonomic Driversof Functional Shifts in the Human Microbiome
这篇文章提出的有关功能上的改变同微生物分类单元之间的关系,运用于人体微生物原理:通过对样品的功能和微生物区系的测定,以微生物基因组参考数据库将样本功能同对应的微生物联系起来,得到功能改变的驱动因子;
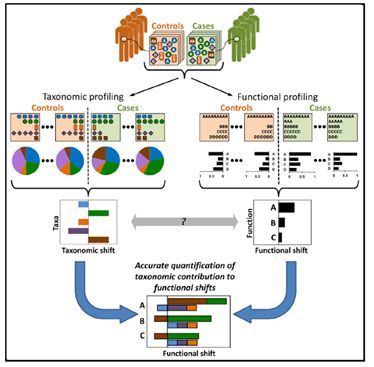
具体算法如下面图所示:首先将微生物丰度数据和功能基因丰度数据输入,功能同微生物关联数据库,(这里我们做环境微生物的,可以使用picrust中的otu同KO关联的文件,也可以使用TAX4FUN中的文件;此处不讨论争议,只介绍方法)输入;通过OTU-ko数据库将OTU数据和KO数据关联,置换检验,LDA判别后得到最终的结果;
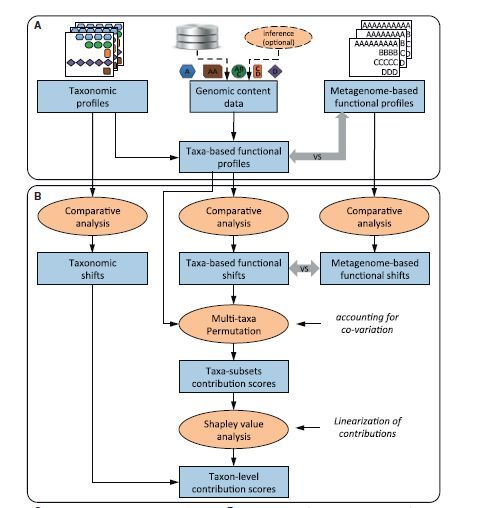
得到最终的效果图的解读:
如D所示:将差异功能转变的驱动因素分为四个部分,使用两个方向的柱状图表示;
1,与处理相关驱动相应功能丰度增加的微生物(右上);
2,与处理相关抑制相应功能丰度增加的微生物(左上);
3,与对照相关驱动相应功能丰度增加的微生物(右下);是这样的,对照中占有重要地位的微生物由于其促进该功能丰度增加的基因很少,因此,这些微生物在对照中的富集会导致对照中此功能相对丰度降低,促进了处理中次功能显著;
4,与对照相关抑制相应功能丰度增加的微生物(左上);也就是这类微生物的此种功能基因含量高;

fishtaco主页:http://borenstein-lab.github.io/fishtaco/index.html

摘要:
1:fishtaco安装起来非常麻烦,之前我一直在使用Qiime和Qiime2两个虚拟Ubuntu系统,在这两个系统中都安装后,发现只能在Qiime中成功安装,可能是python版本问题,官方也只是说过在在python2.7和3.3版本测试过没有问题,可能Qiime2中的python3.6有一些更新是fishtaco无法适应的;
![]()
2:MUSICC标准化似乎要单独做,因为fishtaco提示做MUSICC标准化错误
3:基于picrust数据库做,实在是太慢,到最后我都没能跑出来,只能将其缩小,这里我采用了R语言操作,但是还是够笨的了,希望大家有优化
4:single_taxa方法是在是快得多,可以使用这个方法先看看效果;
5:我们通过fishtaco得到的仅仅是与处理相关的功能富集通路,想到与对照相关的通路,跟换分组标签即可,同时我们可以仅仅跑一个我们关注的通路的分析,节省时间;
现在我来演示安装fishaco:基于Qiime19.1安装的,其实我应该使用Qiime2来做这些工作的,因为python3.6的原因,很多之前开发的脚本无法直接在python3.6上运行,因此Qiime1我还是保留下来,但是Qiime2已经有conda,无疑将来我会转到Qiime2中;
这些个模块必须同时安装到python中,这里值得注意的是,尽量安装官网上标注的这个版本,因为自这个分析以来,已经一年时间了,有些以来模块跟新频繁,比如scikt-learn目前已经到0.19.3版本了,但是当时作者是在0.15.2版本测试的,经过检测不能安装0.19.3版本,有一个功能fishtaco所依赖的发生了变化;
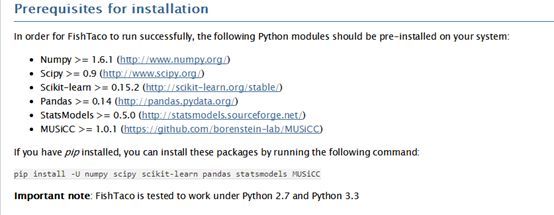
所以下面一条命令就应该这样写了:
#首先安装其依赖的模块
#这里我展示如何安装指定版本的python代码,sudo解决权限问题
sudo pip install -U numpy scipypandas statsmodels MUSiCC
#这里使用pip安装0.15.2版本
sudo pip install"scikit-learn==0.15.2"
如果没有指定镜像源,那么可能会特别慢,可以自行下载后,解压安装,参考下面命令
#解压
tar -xzf fishtaco-1.1.1.tar.gz
#进入目录
cd fishtaco-1.1.1
#安装
python setup.py install
剩下的按照流程即可以成功;
#测试:
test_fishtaco.py
最后显示内容为:OK即为成功;
由此
Qiime1中已近成功安装fishtaco
就picrust做fishtaco,官网有过讨论:http://borenstein-lab.github.io/fishtaco/FAQ.html#can-i-run-fishtaco-with-picrust-derived-metagenomic-functional-profile(点击查看)

下载我们来开始学习:
官网pipline:http://borenstein-lab.github.io/fishtaco/execution.html#usage
就一条命令,十分好学:

这个pipline我们会发现主要需要三个文件:
第一个:就是标准化的otu表格文件
第二个:就是经过MUSICC标准化后的文件
第三个:就是picrust中OTU-KO表格(也就是otu同功能基因关联的文件)
这个分析我们使用16s测序数据,因此我们有otu丰度文件,做一个功能预测,现在我们来做一个picrust我们会得到功能基因文件,直接调用picrust中的OTU-KO文件即为第三个文件:
基于Qiime我们操作:
##这里我自己数据是反向互补的,此时我调整过来,使用seqkit软件,Qiime中没有安装,可以自行安装,小软件很方便;
seqkit seq -p -r seqs_usearch_hg.fa> seqs_usearch_hgfanbu.fa
#转入服务器汇总使用usearch进行处理,没有服务器的可以本地usearch
在我使用服务器的时候发现,每个人是有一定空间的,如果满了就上传不上去了,这个点一定要注意,以后如果发现无法传输,就先看看是否是自己可以存的文件达到上限了
usearch -usearch_global/home/yuanjun/HG_kangbing/seqs_usearch_hgfanbu.fa -db/home/yuanjun/WT/gg_13_5_otus/rep_set/97_otus.fasta -otutabout/home/yuanjun/HG_kangbing/gg135_otu_table.txt -biomout/home/yuanjun/HG_kangbing/g135_table.biom -strand plus -id 0.97 -threads 20

# 转入qiime转换文本为biom标准格式
biom convert -igg135_otu_table.txt -o gg135_otu_table.biom --table-type="OTU table"--to-json
#这个otu表格做一个统计查看一下
biom summarize-table -i gg135_otu_table.biom#序列数很多

# 按拷备数标准化OTU表, -c指定数据库
normalize_by_copy_number.py -i gg135_otu_table.biom-o gg135_otu_table_normalized_otus.biom -c~/Desktop/picrust/picrust/data/16S_13_5_precalculated.tab.gz
#没有基因的名字
predict_metagenomes.py -igg135_otu_table_normalized_otus.biom -o metagenome_predictions.biom -c~/Desktop/picrust/picrust/data/ko_13_5_precalculated.tab.gz
# 转换结果为txt查看
biom convert -imetagenome_predictions.biom -o metagenome_predictions.txt--table-type="OTU table" --to-tsv
metagenome_predictions.txt:结果文件后面没有基因注释
# 调整为标准表格
sed -i '/# Const/d;s/#OTU //g'metagenome_predictions.txt
基因文件我们就得到了
下面我们来进一步得到fishtaco所需要的文件:
下载这个数据库,我在本地已经有了,直接调用即可,没有的去下载:http://picrust.github.io/picrust/picrust_precalculated_files.html#id1(我下载KO数据库,可以选择其他数据)


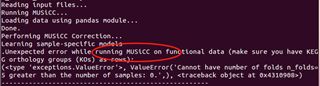
文件准备到了,下面我们应该做标准化了,基因做标准化我想直接在运行过程中进行,但是出错了;
下面我来就otu表格和基因表格做标准化处理:
首先挑去100个丰度最大的otu
biom convert -i~/Desktop/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/gg135_otu_table.txt-o gg135_otu_table.biom --table-type="OTU table" --to-json
filter_otus_from_otu_table.py -n1285 -i gg135_otu_table.biom -o otu_table1285.biom
biom summarize-table -i gg135_otu_table.biom
可以看到我前100个呕吐占的序列数已经超过五万条了,占到了原始序列的80%以上,所以可行
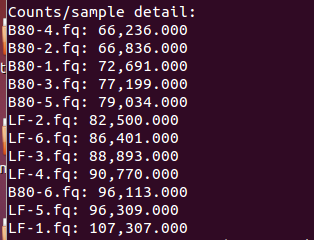
biom convert -iotu_table1285.biom -o otu_table1285.txt --to-tsv
切换到R语言,做otu表格和基因数据的标准化
#############
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/第四次运行fishtaco")
otu = read.table("gg135_otu_table.txt",header=T, row.names= 1, sep="\t")
head(otu)
# 转换原始数据为百分比
norm =t(t(otu)/colSums(otu,na=T)) * 100 # normalization to total 100
write.table(norm,"gg13.5_tu_table_norm.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
metagenome =read.table("metagenome_predictions.txt", header=T, row.names= 1,sep="\t")
head(metagenome)
# 转换原始数据为百分比
norm =t(t(metagenome)/colSums(metagenome,na=T)) * 100 # normalization to total 100
write.table(norm,"metagenome_predictions_norm.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
##
转入Qiime,直接使用MUSICC做标准化
#使用musicc标准化基因数据
run_musicc.pymetagenome_predictions_norm.txt -n -perf -v -ometagenome_predictions_MUSiCC_Normalized.txt
完成之后我首先使用软件自带例子跑了一遍,
#我使用官网上面的这个例子
run_fishtaco.py -taMETAPHLAN_taxa_vs_SAMPLE_for_K00001.tab -fuWGS_KO_vs_SAMPLE_MUSiCC_only_K00001.tab -l SAMPLE_vs_CLASS.tab -gcMETAPHLAN_taxa_vs_KO_only_K00001.tab -op fishtaco_out_no_inf -map_function_levelnone -functional_profile_already_corrected_with_musicc -assessment single_taxa–log
参数解释
-op fishtaco_out_no_inf:设定输出结果文件前缀
-map_function_level none:将功能基因映射到路径,这里设置为无,之前不能跑的时候设置为pathway,但是当我更换为pathway的时候发现其可以运行,因此这也不是最主要的
-functional_profile_already_corrected_with_musicc:表示在运行FishTaco之前,功能配置文件已被MUSiCC更正
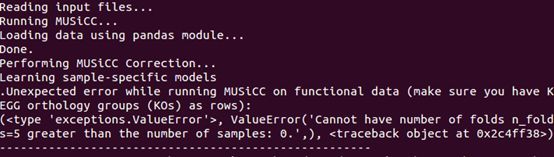
-assessment single_taxa :single_taxa运行时间显著低于multi_taxa,但是准确性低,发现文章上使用的是:multi_taxa
-log:写入日志文件
###接下来我自己的数据跑一下
run_fishtaco.py -tagg135_otu_table_100ge.txt -fu metagenome_predictions1ceshi.txt -lbreak_mapping.txt -gc ~/Desktop/picrust/picrust/data/ko_13_5_precalculated.tab.gz-op fishtaco_out_no_inf -map_function_level none-functional_profile_already_corrected_with_musicc -assessment single_taxa -log
进过测试发现还时无法运行:看来目前的问题在于参考基因组过大,也就是存在于picrust的那个文件太大了,需要根据实际情况将其缩小
这个文件5900多列但是有20万行,因此比较大
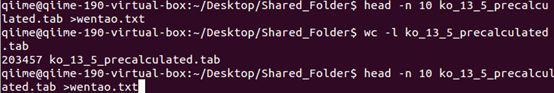
现在我尝试的方法就是将这个文件分割成10个小的文件,逐个与otu表格比对,拿到OTU-KO表格之后再将其合并操作
这一步我采用了本办法,因为picrust那个KO_OTU文件解压之后又5G以上,R语言中打开都是个问题,此时还可以尝试通过逐行读入文件,但是我没用,害怕弄错,电脑死掉;
####在Qiime中操作,将其分隔成11个子文件
head -n 20000ko_13_5_precalculated.tab >~/Desktop/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/wentao2.txt####中间20001-40000行我使用sed命令,重定向到wentao2.txt中
sed -n '20001,40000p'~/Desktop/Shared_Folder/ko_13_5_precalculated.tab > wentao2.txt
##下面我将所有命令一次性编号,然后开始同时分割
sed -n '40001,60000p'~/Desktop/Shared_Folder/ko_13_5_precalculated.tab > wentao3.txt
sed -n '60001,80000p'~/Desktop/Shared_Folder/ko_13_5_precalculated.tab > wentao4.txt
sed -n '80001,100000p'~/Desktop/Shared_Folder/ko_13_5_precalculated.tab > wentao5.txt
sed -n '100001,120000p'~/Desktop/Shared_Folder/ko_13_5_precalculated.tab > wentao6.txt
sed -n '120001,140000p'~/Desktop/Shared_Folder/ko_13_5_precalculated.tab > wentao7.txt
sed -n '140001,160000p'~/Desktop/Shared_Folder/ko_13_5_precalculated.tab > wentao8.txt
sed -n '160001,180000p'~/Desktop/Shared_Folder/ko_13_5_precalculated.tab > wentao9.txt
sed -n '180001,195000p'~/Desktop/Shared_Folder/ko_13_5_precalculated.tab > wentao10.txt
sed -n '195001,203457p'~/Desktop/Shared_Folder/ko_13_5_precalculated.tab > wentao11.txt
转入R
####在R中操作将其与otu表格为索引,将其过滤
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/breakOTU_KO_t
#####进入R中开始操作
#开始挑选OTU-KO表格111111111111111
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/breakOTU_KO_table")
picrust =read.table("wentao1.txt", header=T, row.names= 1, sep="\t")
head(picrust)
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input")
otu_table =read.table("gg135_otu_table.txt",row.names= 1,header=T,sep="\t",check.names=F)
#过滤数据并且排序
idx = rownames(picrust) %in%rownames(otu_table)
picrust1 = picrust[idx,]
head(picrust1)
write.table(picrust1,"OTU-KO_1.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
#########清理R中所有的变量
rm(list=ls())
#######继续开始#####22222222222222
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/breakOTU_KO_table")
picrust =read.table("wentao2.txt", header=T, row.names= 1, sep="\t")
head(picrust)
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input")
otu_table =read.table("gg135_otu_table.txt",row.names= 1,header=T,sep="\t",check.names=F)
#过滤数据并且排序
idx = rownames(picrust) %in%rownames(otu_table)
picrust1 = picrust[idx,]
head(picrust1)
write.table(picrust1,"OTU-KO_2.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
###########
rm(picrust)
rm(picrust1)
rm(idx)
############333333333333333后面我将命令整合在一起
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/breakOTU_KO_table")
picrust =read.table("wentao3.txt", header=T, row.names= 1, sep="\t")
head(picrust)
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input")
#过滤数据并且排序
idx = rownames(picrust) %in%rownames(otu_table)
picrust1 = picrust[idx,]
#head(picrust1)
write.table(picrust1,"OTU-KO_3.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
#############
rm(picrust)
rm(picrust1)
rm(idx)
#############4444444444444444
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/breakOTU_KO_table")
picrust =read.table("wentao4.txt", header=T, row.names= 1, sep="\t")
head(picrust)
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input")
#过滤数据并且排序
idx = rownames(picrust) %in%rownames(otu_table)
picrust1 = picrust[idx,]
#head(picrust1)
write.table(picrust1,"OTU-KO_4.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
#############
rm(picrust)
rm(picrust1)
rm(idx)
###########55555555555555555555555
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/breakOTU_KO_table")
picrust =read.table("wentao5.txt", header=T, row.names= 1, sep="\t")
head(picrust)
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input")
#过滤数据并且排序
idx = rownames(picrust) %in%rownames(otu_table)
picrust1 = picrust[idx,]
#head(picrust1)
write.table(picrust1,"OTU-KO_5.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
#############
rm(picrust)
rm(picrust1)
rm(idx)
###########666666666666666
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/breakOTU_KO_table")
picrust =read.table("wentao6.txt", header=T, row.names= 1, sep="\t")
#head(picrust)
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input")
#过滤数据并且排序
idx = rownames(picrust) %in%rownames(otu_table)
picrust1 = picrust[idx,]
#head(picrust1)
write.table(picrust1,"OTU-KO_6.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
##############
#############
rm(picrust)
rm(picrust1)
rm(idx)
###########777777777777777
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/breakOTU_KO_table")
picrust =read.table("wentao7.txt", header=T, row.names= 1, sep="\t")
#head(picrust)
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input")
#过滤数据并且排序
idx = rownames(picrust) %in%rownames(otu_table)
picrust1 = picrust[idx,]
#head(picrust1)
write.table(picrust1,"OTU-KO_7.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
############
rm(picrust)
rm(picrust1)
rm(idx)
###########888888888888
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/breakOTU_KO_table")
picrust =read.table("wentao8.txt", header=T, row.names= 1, sep="\t")
#head(picrust)
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input")
#过滤数据并且排序
idx = rownames(picrust) %in%rownames(otu_table)
picrust1 = picrust[idx,]
#head(picrust1)
write.table(picrust1,"OTU-KO_8.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
###########
rm(picrust)
rm(picrust1)
rm(idx)
###########99999999999
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/breakOTU_KO_table")
picrust =read.table("wentao9.txt", header=T, row.names= 1, sep="\t")
#head(picrust)
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input")
#过滤数据并且排序
idx = rownames(picrust) %in%rownames(otu_table)
picrust1 = picrust[idx,]
#head(picrust1)
write.table(picrust1,"OTU-KO_9.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
##############
rm(picrust)
rm(picrust1)
rm(idx)
###########10101011011010101010101010101001010
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/breakOTU_KO_table")
picrust =read.table("wentao10.txt", header=T, row.names= 1,sep="\t")
#head(picrust)
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input")
#过滤数据并且排序
idx = rownames(picrust) %in%rownames(otu_table)
picrust1 = picrust[idx,]
#head(picrust1)
write.table(picrust1,"OTU-KO_10.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
##########11 11 11 11 11 11 11
rm(picrust)
rm(picrust1)
rm(idx)
###########10101011011010101010101010101001010
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/breakOTU_KO_table")
picrust =read.table("wentao11.txt", header=T, row.names= 1,sep="\t")
#head(picrust)
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input")
#过滤数据并且排序
idx = rownames(picrust) %in%rownames(otu_table)
picrust1 = picrust[idx,]
#head(picrust1)
write.table(picrust1,"OTU-KO_11.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
##########
下面就是合并了
#############下面开始合并数据框#############
setwd("E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input")
OK0<-read.table("wentaoheadname.txt", header=T, row.names= 1,sep="\t")
OK1<-read.table("OTU-KO_1.txt", header=T, row.names= 1,sep="\t")
colnames(OK1)=colnames(OK0)
head(OK1)
OK2<-read.table("OTU-KO_2.txt", header=T, row.names= 1,sep="\t")
colnames(OK2)=colnames(OK0)
bind<-rbind(OK1,OK2)
head(bind)
##########
OK3 = read.table("OTU-KO_3.txt",header=T, row.names= 1, sep="\t")
colnames(OK3)=colnames(OK0)
OK4 =read.table("OTU-KO_4.txt", header=T, row.names= 1,sep="\t")
colnames(OK4)=colnames(OK0)
bind2<-rbind(OK3,OK4)
##########
OK5 =read.table("OTU-KO_5.txt", header=T, row.names= 1,sep="\t")
colnames(OK5)=colnames(OK0)
OK6 =read.table("OTU-KO_6.txt", header=T, row.names= 1,sep="\t")
colnames(OK6)=colnames(OK0)
bind3<-rbind(OK5,OK6)
############
OK7 =read.table("OTU-KO_7.txt", header=T, row.names= 1,sep="\t")
colnames(OK7)=colnames(OK0)
OK8 =read.table("OTU-KO_8.txt", header=T, row.names= 1,sep="\t")
colnames(OK8)=colnames(OK0)
bind4<-rbind(OK7,OK8)
#############
OK9 =read.table("OTU-KO_9.txt", header=T, row.names= 1,sep="\t")
colnames(OK9)=colnames(OK0)
OK10 =read.table("OTU-KO_10.txt", header=T, row.names= 1,sep="\t")
colnames(OK10)=colnames(OK0)
bind5<-rbind(OK9,OK10)
##########
bind6<-rbind(bind,bind2)
bind7<-rbind(bind3,bind4)
OK11 =read.table("OTU-KO_11.txt", header=T, row.names= 1,sep="\t")
colnames(OK11)=colnames(OK0)
bind8<-rbind(bind5,OK11)
############
bind9<-rbind(bind6,bind7)
###zuizhong
bind10<-rbind(bind9,bind8)
#########
write.table(bind10,"OTU-KO.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
bind10$metadata_NSTI<-NULL
write.table(bind10,"OTU-KO_nometadata_nsti.txt",quote= FALSE,row.names = T,
col.names = T,sep = "\t")
###########
rm(list=ls())
完成之后,我来进入Qiime中开始跑fishtaco
#########开始正式运行
#开始跑,注意我使用的是single_taxa模式,此模式不够准确,但是很快,下面是官网讨论

run_fishtaco.py -taotu_table1285_norm.txt -fu metagenome_predictions_MUSiCC_Normalized.txt -lbreak_mapping.tab -gc OTU-KO_nometadata_nsti.txt -op fishtaco_out_no_inf-map_function_level none -functional_profile_already_corrected_with_musicc-assessment single_taxa -log

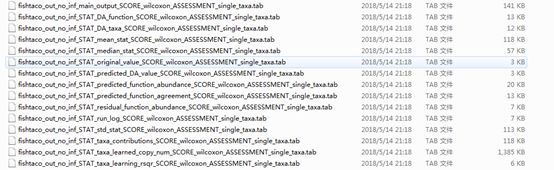
已经出现了结果:如下所示
现在参考官网进行结果解读:
fishtaco_out_no_inf_main_output_SCORE_wilcoxon_ASSESSMENT_single_taxa.tab:是主要的输出文件
a: case-associated taxon driving enrichmentof function in cases; 与处理相关的功能基因富集
b: case-associated taxon attenuatingenrichment of function in cases; 与处理相关的功能基因降低
c: control-associated taxon drivingenrichment of function in cases; 与CK相关的功能基因富集
d: control-associated taxon attenuatingenrichment of function in cases; 与CK相关的功能基因降低

此时我们在加上一个注释otu的文件即可:

下面我们来做可视化,参考网址:http://borenstein-lab.github.io/fishtaco/visualization.html
提到两种方法,可以使用网站,也可以使用R,我选择了R:

现在我来进入R做一个可视化:
##首先我定义了许多颜色:##
mi=c("#E41A1C","#377EB8", "#4DAF4A", "#984EA3","#FF7F00" ,"#FFFF33", "#7c260b",
"#1B9E77", "#D95F02","#7570B3" ,"#E7298A", "#66A61E","#E6AB02",
"#8DD3C7", "#FFFFB3","#BEBADA","#FB8072" ,"#80B1D3","#FDB462", "#B3DE69",
"#FCCDE5","#D9D9D9","#BC80BD","grey60","#E7278A","#64A61E")
p<-MultiFunctionTaxaContributionPlots(input_dir="E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/第三次fishtacomulti_taxa", input_prefix="fishtaco_out_no_inf",input_taxa_taxonomy="E:/Shared_Folder/HG_kangbing/nobac_noqianheti_chuli/fishtaco_input/第三次fishtacomulti_taxa/tax_100.txt",sort_by="list", plot_type="bars", input_function_filter_list=c("ko00130","ko00195","ko00196","ko00785","ko00860","ko00906","ko00941", "ko00945", "ko00984", "ko01053","ko03010" ,"ko03020", "ko04973"), add_predicted_da_markers=F, add_original_da_markers=F,input_permutation="single_taxa ",
#参数简单介绍
input_dir=:输入文件路径
input_prefix:文件名的前缀
input_taxa_taxonomy=:otu的注释文件
sort_by="list",对基因进行排序,按照list
plot_type="bars", 做成柱状图
input_function_filter_list=c("ko00020","ko00540","ko02040"),:这就是功能排序列表,这里有很多的通路,但是只展示其中的几个
add_predicted_da_markers=TRUE,根据分类添加菱形标记
add_original_da_markers=TRUE:添加基于宏基因组的方块标记
###对标签以及主题的修改我们就不多说,毕竟是基于gplot的出图,参数都是一样的改;
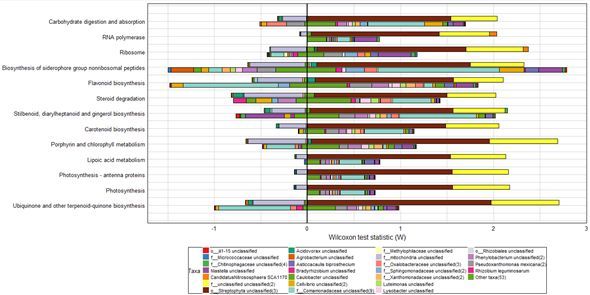
p <- p <- p +scale_x_continuous(breaks=c(1, 2, 3,4,5,6,7,8,9,10,11,12,13),labels=c("Ubiquinone and other terpenoid-quinone biosynthesis",
"Photosynthesis",
"Photosynthesis- antenna proteins",
"Lipoic acid metabolism",
"Porphyrin and chlorophyll metabolism",
"Carotenoid biosynthesis",
"Flavonoid biosynthesis",
"Stilbenoid, diarylheptanoid and gingerol biosynthesis",
"Steroid degradation",
"Biosynthesis of siderophore group nonribosomal peptides",
"Ribosome",
"RNA polymerase",
"Carbohydrate digestion and absorption")) +
guides(fill=guide_legend(nrow=7)) + ylab("Wilcoxon test statistic(W)") +
theme(plot.title=element_blank(),axis.title.x=element_text(size=12,colour="black",face="plain"),
axis.text.x=element_text(size=10,colour="black",face="plain"),axis.title.y=element_blank(),
axis.text.y=element_text(size=10,colour="black",face="plain"),axis.ticks.y=element_blank(),
axis.ticks.x=element_blank(),panel.grid.major.x = element_line(colour="light gray"),panel.grid.major.y = element_line(colour="light gray"),
panel.grid.minor.x =element_line(colour="light gray"), panel.grid.minor.y =element_line(colour="light gray"),
panel.background =element_rect(fill="transparent",colour=NA), panel.border =element_rect(fill="transparent",colour="black"),
legend.background=element_rect(colour="black"),legend.title=element_text(size=10),legend.text=element_text(size=8,face="plain"),
legend.key.size=unit(0.8,"line"),legend.margin=unit(0.1,"line"), legend.position="bottom")
这就是成图:我们可以选择自己关注的通路来做可视化
展示二:
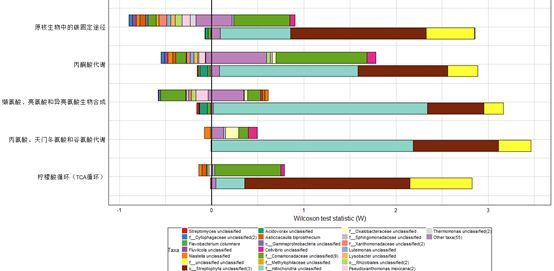
我接下来尝试使用multi_taxa跑一次
######
run_fishtaco.py -ta otu_table1285_norm.txt-fu metagenome_predictions_MUSiCC_Normalized.txt -l break_mapping.tab -gcOTU-KO_nometadata_nsti.txt -op fishtaco_out_no_inf -map_function_level pathway-functional_profile_already_corrected_with_musicc -assessment multi_taxa -log
大概需要6个小时:每个通路差不多需要半个小时才可以跑完,
结果

其次,我们做fishtaco得到的是处理这一组富集的通路,但是我们想要得到对照富集的通路,只需将分组文件中的0和1调换过来即可;

学习永无止境,分享永不停歇!
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
