比PCA更好用的监督排序—LDA分析、作图及添加置信-ggord
线性判别分析LDA
线性判别分析,英文Linear Discriminant Analysis, 以下简称LDA。LDA在模式识别领域(比如人脸识别,舰艇识别等图形图像识别领域)中有非常广泛的应用,在生物学大数据研究中同样也有广泛应用,比如前几个月的Sicence封面文章哈扎人菌群研究就使了此方法,因此我们有必要了解下它的算法原理[1]。
LDA的思想
LDA是一种监督学习的降维技术,也就是说它的数据集的每个样本是有类别输出的。这点和PCA不同。PCA是不考虑样本类别输出的无监督降维技术。LDA的思想可以用一句话概括,就是“投影后类内方差最小,类间方差最大”。什么意思呢? 我们要将数据在低维度上进行投影,投影后希望每一种类别数据的投影点尽可能的接近,而不同类别的数据的类别中心之间的距离尽可能的大。
可能还是有点抽象,我们先看看最简单的情况。假设我们有两类数据 分别为红色和蓝色,如下图所示,这些数据特征是二维的,我们希望将这些数据投影到一维的一条直线,让每一种类别数据的投影点尽可能的接近,而红色和蓝色数据中心之间的距离尽可能的大。

图1. LDA排序原理模式
上图提供了两种投影方式,哪一种能更好的满足我们的标准呢?从直观上可以看出,右图要比左图的投影效果好,因为右图的黑色数据和蓝色数据各个较为集中,且类别之间的距离明显。左图则在边界处数据混杂。以上就是LDA的主要思想了,当然在实际应用中,我们的数据是多个类别的,我们的原始数据一般也是超过二维的,投影后的也一般不是直线,而是一个低维的超平面[1]。
更多算法和原理解析,可阅读参考文献[1]。
LDA排序和ggord添加椭圆置信区间
# Installation
devtools::install_github('fawda123/ggord')
# 加载lda包
library(MASS)
# 查看测试数据
head(iris)
# 按物种分组LDA排序
ord <- lda(Species ~ ., iris, prior = rep(1, 3)/3)
# 展示LDA分析
library(ggord)
p <- ggord(ord, iris$Species)
p
图2. 以鸢尾花尺寸属性按品种分组上色,并添加半透明色0.95置信背景椭圆。箭头为花尺寸各属性与LD1/2轴的相关和贡献[4]。
# 计算置信椭圆函数
get_lda_ell <- function(ord_in, grp_in, ellipse_pro = 0.97){
## adapted from https://github.com/fawda123/ggord/blob/master/R/ggord.R
require(plyr)
axes = c('LD1', 'LD2')
obs <- data.frame(predict(ord_in)$x[, axes])
obs$Groups <- grp_in
names(obs)[1:2] <- c('one', 'two')
theta <- c(seq(-pi, pi, length = 50), seq(pi, -pi, length = 50))
circle <- cbind(cos(theta), sin(theta))
ell <- ddply(obs, 'Groups', function(x) {
if(nrow(x) <= 2) {
return(NULL)
}
sigma <- var(cbind(x$one, x$two))
mu <- c(mean(x$one), mean(x$two))
ed <- sqrt(qchisq(ellipse_pro, df = 2))
data.frame(sweep(circle %*% chol(sigma) * ed, 2, mu, FUN = '+'))
})
names(ell)[2:3] <- c('one', 'two')
ell <- ddply(ell, .(Groups), function(x) x[chull(x$one, x$two), ])
ell
}
# 计算置信椭圆,并添加至原图
anotherEll <- get_lda_ell(ord, iris$Species, 0.97)
## Loading required package: plyr
p + geom_polygon(data = anotherEll,
aes_string(color = 'Groups', group = 'Groups'),
lty=2, fill = NA)
图3. 添加0.97置信区间的虚线椭圆
以菌群测试数据实战
# 读入实验设计
design = read.table("design.txt", header=T, row.names= 1, sep="\t")
# 读取OTU表
otu_table = read.delim("otu_table.txt", row.names= 1, header=T, sep="\t")
# 转换原始数据为百分比
norm = t(t(otu_table)/colSums(otu_table,na=T)) * 100 # normalization to total 100
# 按mad值排序取前6波动最大的OTUs
mad.5 = head(norm[order(apply(norm,1,mad), decreasing=T),],n=6)
row.names(mad.5)=c("Streptophyta","Rubrivivax","Methylibium","Streptosporangiaceae","Streptomyces","Niastella")
data=as.data.frame(t(mad.5))
# 添加分组信息
data$group=design[row.names(data),]$genotype
# 按实验基因组分组排序
ord <- lda(group ~ ., data)
# 使用ggbiplot展示lda(可选)
library(ggbiplot)
ggbiplot(ord, obs.scale = 1, var.scale = 1,
groups = data$group, ellipse = TRUE,var.axes = F)
# 展示LDA分析
library(ggord)
p <- ggord(ord, data$group, ellipse_pro = 0.68)
p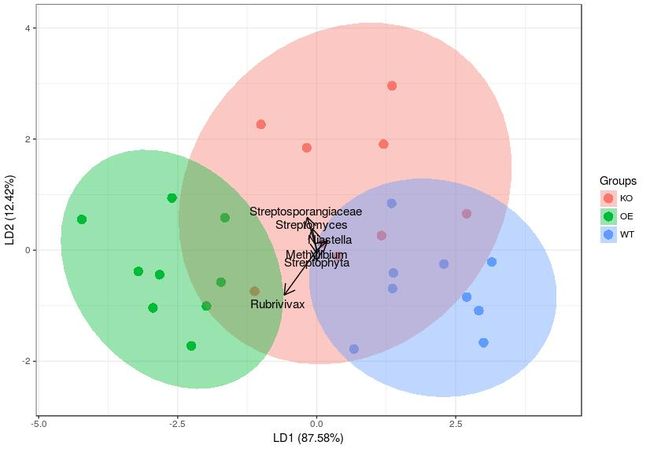
图4. LDA分析三组6个高度差异菌的分组+背景置信区间
anotherEll <- get_lda_ell(ord, data$group, 0.97)
p + geom_polygon(data = anotherEll,
aes_string(color = 'Groups', group = 'Groups'),
lty=2, fill = NA)
图4. LDA分析三组6个高度差异菌的分组+背景置信区间。按68%和97%天加置信区间背景色和虚线椭圆
LDA与PCA比较
LDA用于降维,和PCA有很多相同,也有很多不同的地方,因此值得好好的比较一下两者的降维异同点。
首先我们看看相同点:
1)两者均可以对数据进行降维。
2)两者在降维时均使用了矩阵特征分解的思想。
3)两者都假设数据符合高斯分布。
我们接着看看不同点:
1)LDA是有监督的降维方法,而PCA是无监督的降维方法
2)LDA降维最多降到类别数k-1的维数,而PCA没有这个限制。
3)LDA除了可以用于降维,还可以用于分类。
4)LDA选择分类性能最好的投影方向,而PCA选择样本点投影具有最大方差的方向。
这点可以从下图形象的看出,在某些数据分布下LDA比PCA降维较优。

LDA算法小结
LDA算法既可以用来降维,又可以用来分类,但是目前来说,主要还是用于降维。在我们进行实验数据组间差异分析时,LDA是一个有力的工具。下面总结下LDA算法的优缺点。
LDA算法的主要优点有:
1)在降维过程中可以使用类别的先验知识经验,而像PCA这样的无监督学习则无法使用类别先验知识。
2)LDA在样本分类信息依赖均值而不是方差的时候,比PCA之类的算法较优。
LDA算法的主要缺点有:
1)LDA不适合对非高斯分布样本进行降维,PCA也有这个问题。
2)LDA降维最多降到类别数k-1的维数,如果我们降维的维度大于k-1,则不能使用LDA。当然目前有一些LDA的进化版算法可以绕过这个问题。
3)LDA在样本分类信息依赖方差而不是均值的时候,降维效果不好。
4)LDA可能过度拟合数据。[1]
Reference
- 线性判别分析LDA原理总结 - 刘建平Pinard - 博客园 https://www.cnblogs.com/pinard/p/6244265.html
- 本文方法主要参考Chenhao的博客 http://lchblogs.netlify.com/post/2017-12-22-r-addconfellipselda/
- 《扩增子分析教程-3统计绘图-冲击高分文章》http://mp.weixin.qq.com/s/6tNePiaDsPPzEBZjiCXIRg
- ggbiplot-最好看的PCA作图:样品PCA散点+分组椭圆+变量贡献和相关
- ggord主页 https://github.com/fawda123/ggord
猜你喜欢
- 一文读懂:1微生物组 2进化树 3预测群落功能
- 热文:1图表规范 2DNA提取 3 实验vs分析
- 必备技能:1提问 2搜索 3Endnote
- 文献阅读 1热心肠 2SemanticScholar 3geenmedical
- 扩增子分析:1图表解读 2分析流程 3统计绘图 4群落功能 5进化树
- 科研团队经验:1云笔记 2云协作 3公众号
- 系列教程:1Biostar 2微生物组 3宏基因组
- 生物科普 1肠道细菌 2生命大跃进 3细胞的暗战 4人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外100+ PI,900+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”

点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA