PhiSpy:在细菌基因组中识别噬菌体
PhiSpy:在细菌基因组中识别噬菌体
PhiSpy: a novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies
原文链接:https://doi.org/10.1093/nar/gks406
Nucleic Acids Research [IF: 11.140]
Resource: 2012-05-14
介绍
溶源噬菌体是将自身的基因整合到宿主细菌基因组中,并作为宿主细菌基因组的一部分进行复制。这些可移动的元素可以对它们的细菌宿主的基因组和表型产生巨大的影响,这可能增加菌株的多样性,增加毒性或抗生素抗性。

PhiSpy是一个在细菌(或者古菌)基因组中识别溶源噬菌体的工具。输入一个经过注释的基因组,它会识别出中最可能是噬菌体的区域。PhiSpy的原理是识别出溶源噬菌体的几个显著特征,包括:蛋白质长度,转录链的方向,AT、CG的偏斜性(skew),噬菌体特异words(后面有解释words具体指什么)的丰度,噬菌体的插入位点和噬菌体蛋白的相似性。
In this study, a weighted phage detection algorithm, PhiSpy was developed based on seven distinctive characteristics of prophages, i.e. protein length, transcription strand directionality, customized AT and GC skew, the abundance of unique phage words, phage insertion points and the similarity of phage proteins.
在测试数据集中,可以准确预测94%的溶源噬菌体,假阴性率为6%,假阳性率为0.66%。

方法
收集数据
所有细菌的基因组来源于:Phage Annotation Tools and Methods server,Phantome server。
包含的所有的细菌数据可以在这里查看:https://github.com/linsalrob/PhiSpy/tree/master/tests
计算不同的特征
这一步的目的是计算整个基因组的不同的参数,计算并不依赖于单个基因,而是由一组基因组成的滑动窗口(window)组成。这组基因的数量依据溶源噬菌体基因组的平均基因数量定位40.
蛋白质长度(protein length):噬菌体的蛋白质通常相较于细菌的蛋白质更短。
转录链的方向(transcription strand directionality):在细菌基因组中,通常在其环状DNA中只有一个复制起点,两个复制叉可以独立进行。噬菌体基因沿着基因组聚集,因为它们被组织成共同调节的转录单位。这导致一大群噬菌体基因指向同一个方向,即使它与DNA复制相冲突。
AT、CG的偏斜性(customized AT and GC skew):由于噬菌体和细菌在氨基酸和密码子的使用方面有所不同以及外源DNA的插入,因此AT、CG的偏斜性可以用于区分溶源噬菌体和细菌。
噬菌体特异words的丰度(abundance of unique phage words):
什么是words?
每一个基因被分割成的不重叠的12bp的序列。
Each gene was split into 12 bp non-overlapping words, i.e. four non-overlapping codons.
通过分析发现,来源于噬菌体库的words会在噬菌体基因组中更加频繁地出现。
同源性(homology):
一个包含了40个基因的窗口(window)中,如果至少有10个基因的功能在噬菌体子系统中被描述,则该窗口被认为是溶源噬菌体的窗口,否则它被认为是细菌的窗口。
排序算法
这一步的目的是利用随机森林算法将一个窗口(window)分类为噬菌体的窗口或者细菌的窗口。
随机森林算法为整个基因组的每个窗口(window)生成一个等级,表明这个窗口是由细菌基因还是噬菌体基因组成的。
有5个特征会在不同亲缘关系的基因组中非常不同。如果两个基因组有显著的相似性,那么就会被认为是亲缘关系近的基因组。
在Phantome server中有547条细菌基因组(截止2010年3月),其中分成了亲缘关系较近的19个组,每一个组中都至少有一个细菌基因组中含有经过注释的溶源噬菌体,这一个细菌基因组就用作训练集。对于并不属于任何一个组的细菌基因组,就使用一个通用训练集,其中包含了全部41条含有溶源噬菌体的细菌基因组。
对每个基因进行最后的排序
这一步的目的是为每一个基因提供一个预测的状态,对于基因组中的每一个基因,要么是0(非溶源体基因),要么是1(溶源噬菌体基因)。如果窗口(window)大小是n,每个基因贡献1到n个窗口(前面提过窗口是滑动的)。因此,每个基因的最终等级是通过取该基因参与的窗口的平均等级来衡量的。溶源噬菌体预测状态是从最终排名中计算出来的。如果最终等级大于基因组中任何基因最高等级的一半,则该基因被认为是噬菌体基因;否则,它被认为是细菌基因。
对预测进行评估
最后一步的目的是确定att 区域,并对预测的溶源噬菌体进行评估。
当噬菌体整合到宿主的基因组中时,它们通常被两个att 位点所包围——插入位点两侧的一个短的重复序列。当找到att 位点之后,要对其进行验证。如果att 位点位于初始预测范围内,则计算两个间隙(att L和初始预测开始之间以及att R和初始预测结束之间)中的phage-like蛋白的数量。如果这两个缺口中四分之一的基因的功能属于噬菌体子系统,则初始预测被认为是最终预测,否则att 位点覆盖的区域被认为是最终预测。如果att 位点在初始预测之外,则遵循相同的程序。
在验证att 位点后,通过检查该区域所有蛋白质的功能来评估预测的溶源噬菌体。如果有超过5个蛋白质的功能属于噬菌体子系统或者是未知的,并且phage-like/未知蛋白质的数量至少是预测区域中蛋白质总数的一半,则该区域被认为是潜在的溶源噬菌体。然而,如果一组功能属于噬菌体子系统的蛋白质,在分类步骤中不被认为是可能的潜在的溶源噬菌体,那么该区域也被认为是潜在的前噬菌体。
计算假阳性和假阴性
人工策划的(manually curated)噬菌体子系统被用于评估该方法的准确性。设计了一个包含两步的程序来自动计算溶源噬菌体预测的错误率(对于那些在原始基因组分析论文中没有溶源噬菌体信息的基因组)。
在程序的第一步,真阳性(true positives,TP)和假阳性(false positives,FP)被预测。如果预测区域由至少六个噬菌体蛋白质组成,或者预测区域内50%的蛋白质属于噬菌体子系统或是未知的,则预测区域被认为是TP溶源噬菌体,否则该区域被认为是FP而不是溶源噬菌体(这些限制由经验确定)。
被认为是TPs的溶源噬菌体分为两组:(I)已知的(known)溶源噬菌体——如果该区域含有phage-like蛋白;我们认为可以通过基于相似性的方法进行鉴定,因此将其标记为“已知(known)”和(II)未定义的(undefined)溶源噬菌体——如果该区域没有phage-like蛋白;因此,这不太可能被称为溶源噬菌体。
在程序的第二步,如果有至少六个连续的基因,其功能属于噬菌体子系统,并且该区域被鉴定为潜在的溶源噬菌体,则该区域被认为是假阴性(FN)。然而,在这种情况下,假设的蛋白质被忽略了,因为几个假设的蛋白质的存在不足以预测一个区域为溶源噬菌体区域。
安装
Conda(推荐)
conda install -c bioconda phispyPIP
sudo apt install -y build-essential python3-dev python3-pip
python3 -m pip install --user PhiSpy依赖关系
Python - v 3.4 or later
Biopython - v 1.58 or later
gcc - GNU project C and C++ compiler - version 4.4.1 or later
The Python.h header file. This is included in python3-dev that is available on most systems.
一般的服务器或者Linux系统的电脑上会有Python和gcc,Biopython需要安装一下。Python.h这里小编也不知道是啥意思,我装完之后正常运行了,就没有管这里了。
测试
# 下载Streptococcus pyogenes M1 genome的genbank文件
curl -Lo Streptococcus_pyogenes_M1_GAS.gb https://bit.ly/37qFArb
# 运行PhiSpy
PhiSpy.py -o Streptococcus.phages Streptococcus_pyogenes_M1_GAS.gb结果文件就会在Streptococcus.phages文件夹中。
运行
最简单的用法
PhiSpy.py genbank_file -o output_directorygenbank_file:经过注释的genbank格式的输入文件,可以使用RAST或者PROKKA注释。
output_directory:输出文件所在的文件夹
使用来自HMM的信号
PhiSpy.py genbank_file -o output_directory --phmms hmm_db --threads 4hmm_db 用来搜索基因组编码蛋白质的HMM参考数据库
使用pVOG database当做训练集
wget http://dmk-brain.ecn.uiowa.edu/pVOGs/downloads/All/AllvogHMMprofiles.tar.gz
tar -zxvf AllvogHMMprofiles.tar.gz
cat AllvogHMMprofiles/* > pVOGs.hmm使用pVOGs.hmm替换hmm_db。
制作自己的参考数据集
如果在参考数据集中,跟你感兴趣的细菌的亲缘物种缺失,你可以通过提供至少一个注释了溶源噬菌体蛋白质的单个基因组来制作自己的训练集。这是通过为溶源噬菌体区域内的每个CDS特征添加新的限定符GenBank注释来实现的:/is_phage="1 "。这使得Phispy能够区分来自细菌/噬菌体区域的信号,并制作一个训练集,以便以后在使用随机森林算法进行分类时使用。
PhiSpy.py -o output_directory -k kmer_size -t kmers_type -g groups_file --retrain --phmms hmm_db --threads 4 genome.gb.gzoutput_directory:临时和最终文件会写入的地方kmers_size:默认是12kmers_type:默认是“all”,指的是生产1nt的kmers。groups_file:映射GenBank文件的扩展名和他们将要创建的组的名称, 可以参考/test/group.txt
genome.gb.gz:是一个设置了/is_phage="1" 标志gzip压缩的GenBank文件
结果文件
当--output_choice默认为3时
prophage.tbl
一个被tab分隔为两列的文件,包含了溶源噬菌体的id,contig起始位置终点位置。

prophage_tbl.tsv
也是一个被tab分隔的文件。包含了基因组所有的基因。如果第10列是1,那么就是噬菌体基因,否则是一个细菌基因。
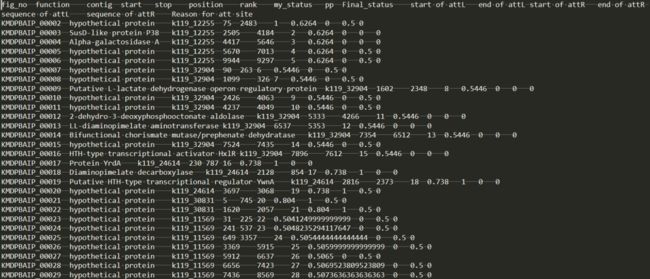
prophage_coordinates.tsv
包含了溶源噬菌体的id,contig,起始位置,终止位置和att区域。
![]()
其他的输出文件
如果希望获取到其他的输出文件,可以用--output_choice设置,将你需要的文件的code数相加。
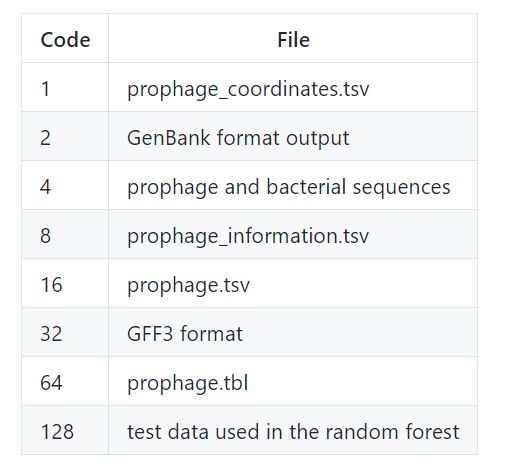
举个例子,如果你想得到GenBank format output(2)和prohage _ information . tsv(8),那么输入--output_choice 10 。
小编有话说
这个软件存在数据量低的问题。软件构建之初只使用了547条细菌基因组,其中分成了亲缘关系较近的19个组,每一个组中至少有一个细菌基因组中含有经过注释的溶源噬菌体,这一个细菌基因组就用作训练集。训练集会不会太少了?真的够吗?
作者还提到,如果在参考数据集中,感兴趣的细菌的亲缘物种缺失,可以通过提供至少一个注释了溶源噬菌体蛋白质的单个基因组来制作自己的训练集。小编不太明白跟细菌物种亲缘关系的远近有什么关系?就小编自己的研究来看,细菌亲缘关系近,也不代表溶源噬菌体的亲缘关系就会近,大概也不会代表在溶源噬菌体基因组上有什么更相似的特点。欢迎各位老师同学一起来讨论~
参考文献
https://github.com/linsalrob/PhiSpy
Akhter S, Aziz R K, Edwards R A. PhiSpy: a novel algorithm for finding prophages in bacterial genomes that combines similarity-and composition-based strategies[J]. Nucleic acids research, 2012, 40(16): e126-e126.
撰文:秋芒树,华西生物医学大数据中心
责编:刘永鑫,中科院遗传发育所
译者简介
秋芒树,本科毕业于中国农业大学,硕士毕业于英国帝国理工学院,现就职于华西生物医药大数据中心。关注婴儿肠道微生物,肠道细菌与噬菌体的相互作用。在宏基因组公众号发表《Nature:TEDDY计划中幼儿肠道微生物组随时间的发育》、《Cell子刊:成年同卵双胞胎的病毒组多样性与肠道微生物组多样性相关》、《CHM:新生儿肠道微生物菌群研究》 等。欢迎批评、指正和交流, 微信:W18813001339.
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读