最终版本Science级组合图表绘制
简介
ggcor 是厚哥最近的作品,功能完全代替了science组合图表绘制。这里我也为大家带力实战教程,总体来说厚哥这个ggcor包用起来还是挺方便的,将许多功能简化,为我们提供了一个很好的入门机会。所以大家也别再私信我要之前两篇推送的原始数据了,这次推送完全替代之前的版本。重要事情说三遍!这是sicence组合图表最终版本别用之前的了!这是sicence组合图表最终版本别用之前的了!这是sicence组合图表最终版本别用之前的了!
这里我献出厚哥的信息:
厚缊 业余的R语言可视化重度患者
个人博客:houyun.xyz
邮箱:[email protected]
ggcor在微生物生态领域的使用初
![]()
目前厚哥这个包还在不断开发中,但是我已经迫不及待想试试了。包的安装使用github安装函数install_github。
简单来说一下这个包用来干什么的?(下面摘抄自厚哥亲笔)
相关系数矩阵可视化已经至少有两个版本的实现了,魏太云基于base绘图系统写了corrplot包,应该说是相关这个小领域中最精美的包了,使用简单,样式丰富,只能用惊艳来形容。Kassambara的ggcorrplot基于ggplot2重写了corrplot,实现了corrplot中绝大多数的功能,但仅支持“square”和“circle”的绘图标记,样式有些单调,不过整个ggcorrplot包的代码大概300行,想学习用ggplot2来自定义绘图函数,看这个包的源代码很不错。还有部分功能相似的corrr包(在写ggcor之前完全没有看过这个包,写完之后发现在相关系数矩阵变data.frame方面惊人的相似),这个包主要在数据相关系数提取、转换上做了很多的工作,在可视化上稍显不足。ggcor的核心是为相关性分析、数据提取、转换、可视化提供一整套解决方案,目前的功能大概完成了70%,后续会根据实际需要继续扩展。
if(!require(devtools))
install.packages("devtools")
if(!require(ggcor))
devtools::install_github("houyunhuang/ggcor")首先学习函数
![]()
待会咱们要使用这些函数完成实战,也挑选出来我喜欢的几个可视化方案。这里的示例数据都是现成的,所以大家在这里对照运行即可。
导入r包和示例数据
suppressWarnings(suppressMessages(library("ggcor")))
corr <- cor(mtcars)
df <- as_cor_tbl(corr)
df ## return a tibble## # A tibble: 121 x 3
## x y r
## *
## 1 1 11 1
## 2 1 10 -0.852
## 3 1 9 -0.848
## 4 1 8 -0.776
## 5 1 7 0.681
## # ... with 116 more rows
## Extra attributes:
## xname: mpg cyl disp hp drat wt qsec vs am gear carb
## yname: carb gear am vs qsec wt drat hp disp cyl mpg
## type: full
## show.diag: TRUE 下面介绍几种本人喜欢的可视化方式,我这里统一将图例修改为离散型颜色:fill.bin = TRUE, legend.breaks = seq(-1, 1, length.out = 5)
注意一下开发版本才能添加颜色设置参数:,fill.bin = TRUE, legend.breaks = seq(-1, 1, length.out = 5) ,文末尾安装方式
#这里不仅仅展示了相关值,而且展示了置信区间,geom_cross用于排除不显著相关。
#
ggcor(mtcars, type = "full", cor.test = TRUE) + geom_confbox()+ geom_cross()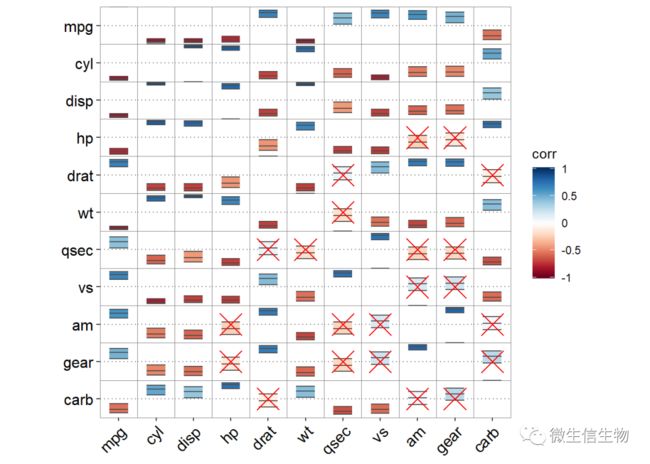 这里使用方形我觉得高档一些,本人比较喜欢,将不显著的去掉。
这里使用方形我觉得高档一些,本人比较喜欢,将不显著的去掉。
# 这里使用方形我觉得高档一些,本人比较喜欢
ggcor(mtcars, type = "full", cor.test = TRUE) +
geom_square() + geom_cross()
配合颜色填充和相关标注,加上显著性标签很容易整体把控指标的相关性。使用r = 这里厚哥添加的五角星,我们来了解一下。
# 这里使用方形我觉得高档一些,本人比较喜欢
ggcor(mtcars, type = "full", cor.test = TRUE) +
# geom_square() +
geom_star(n = 5)+
geom_cross()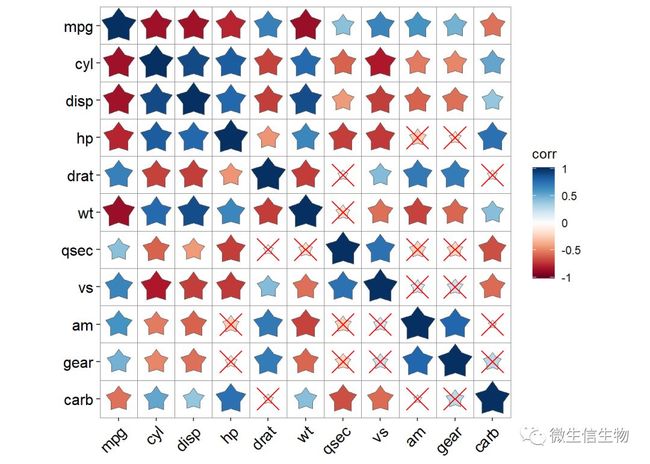 NA参数去除相关值,因为颜色已经表示过相关大小了,值就省略掉吧,mark = "*"参数规范统一的显著性标签。
NA参数去除相关值,因为颜色已经表示过相关大小了,值就省略掉吧,mark = "*"参数规范统一的显著性标签。
#配合颜色填充和相关标注,加上显著性标签很容易整体把控指标的相关性。使用r = NA参数去除相关值,因为颜色已经表示过相关大小了,值就省略掉吧,mark = "*"参数规范统一的显著性标签。
ggcor(mtcars, type = "full", cor.test = TRUE, cluster.type = "all") +
# geom_raster() +
geom_colour()+
geom_mark(r = NA,sig.thres = 0.05, size = 3, colour = "grey90")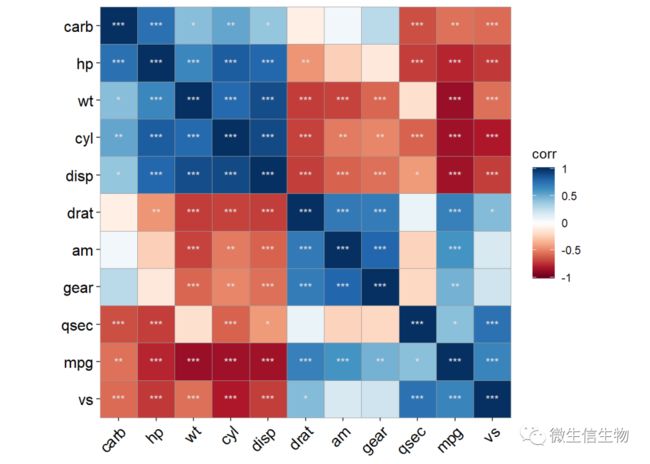 非对称相关图形可以节省空间,很多人曾求助怎么做,其实就是将最后的出图矩阵做相应的裁剪就好了,之前我都是自己裁剪的,现在厚哥包装进去了,方便了很多。
非对称相关图形可以节省空间,很多人曾求助怎么做,其实就是将最后的出图矩阵做相应的裁剪就好了,之前我都是自己裁剪的,现在厚哥包装进去了,方便了很多。
# 非对称相关图形可以节省空间,很多人曾求助怎么做,其实就是将最后的出图矩阵做相应的裁剪就好了,之前我都是自己裁剪的,现在厚哥包装进去了,方便了很多。
suppressWarnings(suppressMessages(library(vegan))) # 使用vegan包所带的数据集
data(varechem)
data(varespec)
df03 <- fortify_cor(x = varechem, y = varespec[ , 1:30], cluster.type = "col")
ggcor(df03) + geom_square() 群落矩阵和环境因子相关
群落矩阵和环境因子相关
corr <- fortify_cor(varechem, type = "upper", show.diag = TRUE,
cor.test = TRUE, cluster.type = "all")
mantel <- fortify_mantel(varespec, varechem,
spec.select = list(spec01 = 22:25,
spec02 = 1:4,
spec03 = 38:43,
spec04 = 15:20),
mantel.fun = "mantel.randtest")
ggcor(corr, xlim = c(-5, 14.5)) +
add_link(mantel, diag.label = TRUE) +
add_diaglab(angle = 45) +
geom_square() + remove_axis("y")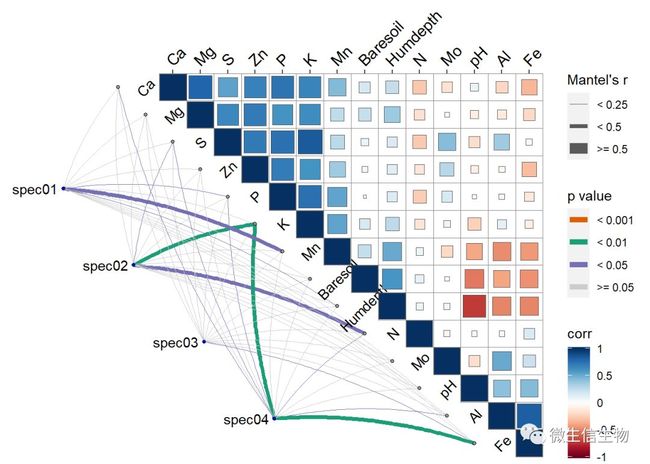
另外一种方式组合
library(cowplot)
mantel <- fortify_mantel(varespec, varechem,
spec.select = list(spec01 = 22:25,
spec02 = 1:4,
spec03 = 38:43,
spec04 = 15:20),
mantel.fun = "mantel.randtest")
mantel$p <- cut(mantel$p, breaks = c(0, 0.001, 0.01, 0.05, 1),
labels = c("< 0.001", "< 0.01", "< 0.05", ">= 0.05"),
right = FALSE, include.lowest = TRUE)
p1 <- ggcor(varechem) + geom_square() + remove_axis("x")
p2 <- ggcor(mantel, mapping = aes(fill = p), is.minimal = TRUE, keep.name = TRUE) +
geom_star(aes(r = 0.65), n = 5, ratio = 0.6) +
scale_fill_manual(values = c("darkgreen", "green", "lightgreen", "grey95"),
drop = FALSE)
plot_grid(p1, p2, ncol = 1, align = "v", labels = c('A', 'B'),
rel_heights = c(0.13*dim( varechem)[2], 1))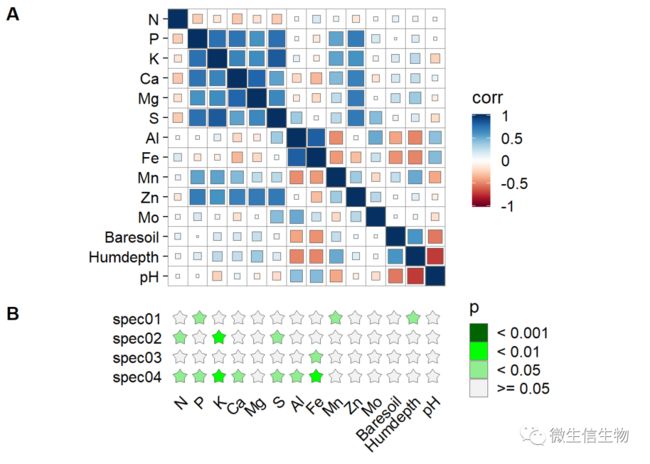
微生物生态学领域实战
![]()
基于phloseq我开发了一系列基于扩增子的数据分析脚本,我也将再不久将这个脚本纳入,这里大家必须学习phyloseq的封装格式和基本用法。
library("phyloseq")
library("vegan")
library("grid")
library("gridExtra")
library("ggplot2")
ps = readRDS(".//ps_OTU_.rds")
ps1 = ps
ps1 = filter_taxa(ps1, function(x) sum(x ) > 200 , TRUE);ps1## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 78 taxa and 24 samples ]
## sample_data() Sample Data: [ 24 samples by 14 sample variables ]
## tax_table() Taxonomy Table: [ 78 taxa by 7 taxonomic ranks ]ps1 = transform_sample_counts(ps1, function(x) x / sum(x) );ps1## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 78 taxa and 24 samples ]
## sample_data() Sample Data: [ 24 samples by 14 sample variables ]
## tax_table() Taxonomy Table: [ 78 taxa by 7 taxonomic ranks ]path = "./phyloseq_5_RDA_CCA_cor/"
dir.create(path)
vegan_otu <- function(physeq){
OTU <- otu_table(physeq)
if(taxa_are_rows(OTU)){
OTU <- t(OTU)
}
return(as(OTU,"matrix"))
}
otu = as.data.frame(t(vegan_otu(ps1)))
mapping = as.data.frame( sample_data(ps1))
env.dat = mapping[,3:ncol(sample_data(ps1))]
env.st = decostand(env.dat, method="standardize", MARGIN=2)#
env_dat = env.st#这里不仅仅展示了相关值,而且展示了置信区间,geom_cross用于排除不显著相关。
ggcor(env_dat, type = "full", cor.test = TRUE) + geom_confbox()+ geom_cross()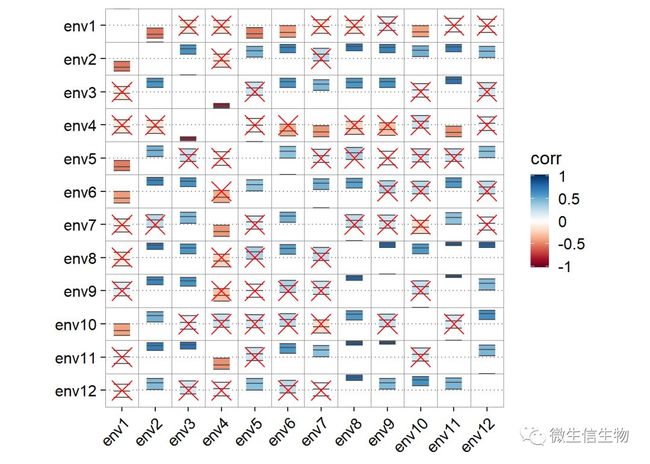 这里使用方形我觉得高档一些,本人比较喜欢,将不显著的去掉。
这里使用方形我觉得高档一些,本人比较喜欢,将不显著的去掉。
# 这里使用方形我觉得高档一些,本人比较喜欢
ggcor(env_dat, type = "full", cor.test = TRUE) +
geom_square() + geom_cross()配合颜色填充和相关标注,加上显著性标签很容易整体把控指标的相关性。使用r = NA参数去除相关值,因为颜色已经表示过相关大小了,值就省略掉吧,mark = "*"参数规范统一的显著性标签。
#配合颜色填充和相关标注,加上显著性标签很容易整体把控指标的相关性。使用r = NA参数去除相关值,因为颜色已经表示过相关大小了,值就省略掉吧,mark = "*"参数规范统一的显著性标签。
ggcor(env_dat, type = "full", cor.test = TRUE, cluster.type = "all") +
# geom_raster() +
geom_colour()+
geom_mark(r = NA,sig.thres = 0.05, size = 3, colour = "grey90")
这样一来我们来挑选微生物和环境因子做相关就方便多了。
# 非对称相关图形可以节省空间,很多人曾求助怎么做,其实就是将最后的出图矩阵做相应的裁剪就好了,之前我都是自己裁剪的,现在厚哥包装进去了,方便了很多。
#计算相关
#太多OTU展示起来不太好看,这里我选择30个展示
ss = t(otu)[,1:30]
df03 <- fortify_cor(x = env_dat, y = ss, cluster.type = "col")
ggcor(df03) + geom_square()
现在我们来组合群落和环境因子关系,这里我模拟了两个群落,这两个群落都是一样的,我通过list收纳这两个群落. xlim = c(-5, (dim(env_dat)[2] +0.5)):在厚哥的指点下修改为环境因子数量加0.5,厚哥表示可能之后这个参数会被写到函数内部。
#转置otu表格,作为第一个群落
otu2 = t(otu)
#同样赋值为第二个群落
otu3 = t(otu)
#无论多少个群落,将其使用list包起来,注意设置名称
spe = list(A = otu2,B = otu3)
#计算环境因子相关
corr <- fortify_cor(env_dat , type = "upper", show.diag = TRUE,
cor.test = TRUE, cluster.type = "all")
#计算环境因子和群落相关
mantel <- fortify_mantel(spe , env_dat,
# spec.select = list(A = 1:dim(otu2)[2]),
mantel.fun = "mantel")
#出图
ggcor(corr, xlim = c(-5,(dim(env_dat)[2] +0.5))) +
add_link(mantel, diag.label = TRUE) +
add_diaglab(angle = 45) +
geom_square() + remove_axis("y")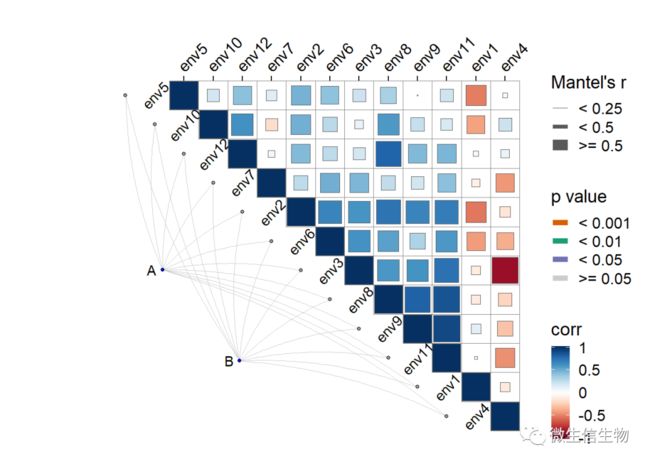 下面展示一个四个群落的数据
下面展示一个四个群落的数据
#转置otu表格,作为第一个群落
otu2 = t(otu)
#同样赋值为第二个群落
otu3 = t(otu)
#无论多少个群落,将其使用list包起来,注意设置名称
spe = list(A = otu2,B = otu3,C = otu3,D = otu3)
corr <- fortify_cor(env_dat , type = "upper", show.diag = TRUE,
cor.test = TRUE, cluster.type = "all")
mantel <- fortify_mantel(spe , env_dat ,
# spec.select = list(A = 1:dim(otu2)[2]),
mantel.fun = "mantel")
ggcor(corr, xlim = c(-5, (dim(env_dat)[2] +0.5))) +
add_link(mantel, diag.label = TRUE) +
add_diaglab(angle = 45) +
# geom_square() +
geom_star(n = 5)+
remove_axis("y")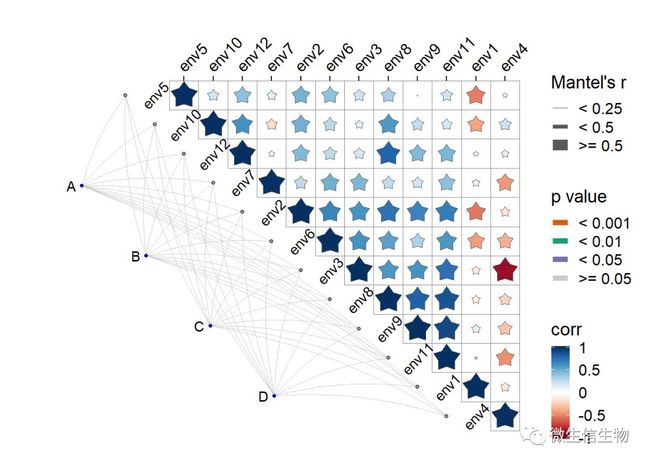
另外一种可视化群落和环境因子关系的组合图表,B图需要调整大小,这里我本来以为是线性映射,做一个变量,显然不是,我还是老老实实修改了一下大小。这里我们的群落和环境因子之间没有相关关系
library(cowplot)
#转置otu表格,作为第一个群落
otu2 = t(otu)
#同样赋值为第二个群落
otu3 = t(otu)
#无论多少个群落,将其使用list包起来,注意设置名称
spe = list(A = otu2,B = otu3,C = otu3,D = otu3)
corr <- fortify_cor(env_dat , type = "upper", show.diag = TRUE,
cor.test = TRUE, cluster.type = "all")
mantel <- fortify_mantel(spe , env_dat ,
# spec.select = list(A = 1:dim(otu2)[2]),
mantel.fun = "mantel")
mantel$p <- cut(mantel$p, breaks = c(0, 0.001, 0.01, 0.05, 1),
labels = c("< 0.001", "< 0.01", "< 0.05", ">= 0.05"),
right = FALSE, include.lowest = TRUE)
p1 <- ggcor(env_dat) +
# geom_square() +
geom_star(n = 5)+
remove_axis("x")
p2 <- ggcor(mantel, mapping = aes(fill = p), is.minimal = TRUE, keep.name = TRUE) +
geom_star(aes(r = 0.65), n = 5, ratio = 0.6) +
scale_fill_manual(values = c("darkgreen", "green", "lightgreen", "grey95"),
drop = FALSE)
plot_grid(p1, p2, ncol = 1, align = "v", labels = c('A', 'B'),
rel_heights = c(0.155*dim(env_dat)[2], 1))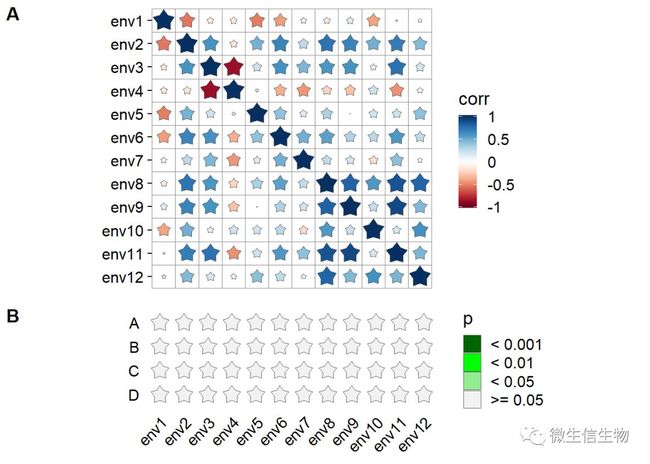
后台回复"ggcor"获得数据和代码下载链接
常见问题
![]()
binned_scale找不到
这是由于ggplot版本问题,这里需要升级成新版本,来自github。
install.packages(“remotes”) remotes::install_github(“hadley/ggplot2”)
Error: ‘decode_colour’ is not an exported object from ‘namespace:farver’
这是farver版本问题
install.packages(“farver”)
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读