随手“一片”SCI,Qiime2扩增子处理流程确定不了解一下?
文章目录
- conda安装qiime2
- 导入数据
- 制作Manifest和Metadata表
- Import数据
- 查看原始数据质量
- DADA2去噪、去嵌合体和生成OTU
- 构建进化树
- 绘制稀释曲线
- 计算物种多样性
- 物种组成分析
- 基于sklearn方法
- 基于blast和vsearch方法
- qiime2其他常用操作
- 导出制表符分割的特征表
- 折叠物种分类单元
- 按样本筛选特征表
- 参考
作者:余涛
email:[email protected]
中国科学院大学
现如今,测序已经将近实现‘千元基因组’,近百元就能测一个扩增子样本,产出数据能广泛揭示不同环境样本的物种组成和丰度等众多信息。扩增子测序就像下饭菜,大米饭里拌几勺吃起来就美味而可口,随手“一片”SCI,Qiime2扩增子处理流程确定不了解一下?
conda安装qiime2
本教程介绍在Linux上的qiime2 命令行版q2cli(qiime2 command line interface),最新版为qiime2-2020.2。安装方式采用最便捷的conda安装方法:
# 下载qiime2的conda安装配置文件
wget https://data.qiime2.org/distro/core/qiime2-2020.2-py36-linux-conda.yml --no-check-certificate
# 创建一个名为 qm2-2020.2的环境,按照qiime2文件安装
conda env create -n qm2-2020.2 --file qiime2-2020.2-py36-linux-conda.yml
# 安装完成后,可删除源文件
rm qiime2-2020.2-py36-linux-conda.yml
# 激活环境
source activate qiime2
# 可查看版本
qiime --version
# 查看更为详尽的配置信息
qiime info

导入数据
理论上qiime2可以从任何一步分析的数据导入进行重新分析,这里以最为常见的公司给的拆分过barcode的pair-end带质量信息的fastq序列示例。(目前,qiime仅能拆分单端数据,对于双端数据的拆分需要自己撸代码或者借助其他工具,暂且不表。)
制作Manifest和Metadata表
在导入数据之前需要制作两个文件:
manifest表用于显示样本名称和对应的双端序列路径
metadata表用于记录样本对应的元数据信息(例如分组、年龄、性别等等)
manifest示例manifest.tsv:

第一列样本名称,第二列为forward序列路径,第三列为reverse序列路径
metadata示例metadata.tsv:

第二列重复样本名称是便于后面显示每个样本的alpha多样性曲线。
- 注意,这两个表都是制表符分割,制作好后可以先在google浏览器插件Keemei上验证文件格式是否标准:
Add ones-Keemei-Validate qiime2 metadata file

验证通过示例:

Import数据
qiime2要求规范数据格式,主要是.qza的数据文件和.qzv的可视化文件,.qza的文件可以通过unzip(推荐)和export方式解压查看,.qzv的文件可通过View qiime2查看。
import的目的就是生成一个标准的包含原始测序数据的.qza文件(官网称之为Artifact)
# import
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \ #生成数据的类型,可使用--show-importable-types查看所有类型
--input-path manifest.tsv \
--output-path paired-end-demux.qza \
--input-format PairedEndFastqManifestPhred33V2 \ #导入数据类型
# 如果样本达数百以上,导入需要一定时间,需要nohup起来,并且可以记录运行时间。这样跑起来以后你就可以安心吃鸡、农药、打豆豆whatever。
{ nohup /usr/bin/time -f "%E" qiime tools import --type 'SampleData[PairedEndSequencesWithQuality]' --input-path Bacteria_PE33_manifest.tsv --output-path paired-end-demux.qza --input-format PairedEndFastqManifestPhred33V2; } &>import.log &
一旦导入数据生.qza的文件,你就可以开始qiime2流程的各种分析,首先查看 原始数据测序质量。
查看原始数据质量
qiime demux summarize \
--i-data paired-end-demux.qza \ #上一步结果
--o-visualization demux-raw-summary.qzv
在Overview可以看到所有样本reads数的基本统计信息:最小、最大值,中位数、平均数。本次分析样本reads数多数在77551附近。

在Interactivate quality plot可以看到双端序列的碱基质量boxplot图,黑框代表50%的数据情况,在3端末尾质量有所下降,后续可视情况去除。

DADA2去噪、去嵌合体和生成OTU
qiime dada2 denoise-paired \
--i-demultiplexed-seqs paired-end-demux.qza \
--p-trunc-len-f 0 \ #截掉3端forward序列X位置后的序列
--p-trunc-len-r 0 \ #截掉3端reverse序列X位置后的序列
--p-n-threads 30 \ #线程
--o-table table.qza \ #FeatureTable[Frequency]
--o-representative-sequences rep-seqs.qza \ #FeatureData[Sequence],每一个feature对应的一个joined的双端代表序列
--o-denoising-stats denoising-stats.qza \
#嵌合体等统计信息
# 可视化统计信息
qiime metadata tabulate \
--m-input-file denoising-stats.qza \
--o-visualization denoising-stats.qzv
现在最重要的OTU特征表和代表序列已经生成,可以通过unzip解压到指定目录unzip rep-seqs.qza -d OTU/ && unzip table.qza -d OTU/,解压后feature-table.biom和dna-sequences.fasta即是特征表和代表序列。
dna-sequences.fasta:
feature-table.biom需要经过biom转换成文本查看:
biom convert -i feature-table.biom -o feature-table.tsv --to-tsv
feature-table.ts,第一行为注释,第二行为样本名称,第三行起为OTU的丰度,每一个OTU ID都可以在代表性序列中找到。
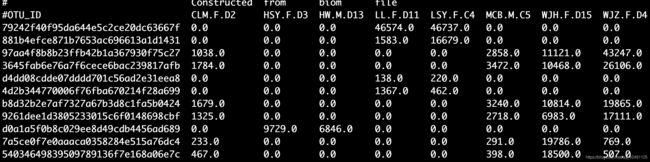
打开denoising-stats.qzv可看到不同步骤处理后剩余的reads,并且可以排序,这里看到的最少的非嵌合体序列也在35000以上,一般满足分析了。

构建进化树
接下来利用代表序列构建进化树,用于计算物种多样性
qiime phylogeny align-to-tree-mafft-fasttree \ #使用mafft比对的fasttree构建进化树
--i-sequences rep-seqs.qza \
--o-alignment aligned-rep-seqs.qza \ #输出的对齐序列
--o-masked-alignment masked-aligned-rep-seqs.qza \#屏蔽掉高可变的复杂序列
--o-tree unrooted-tree.qza \#输出的无根树
--o-rooted-tree rooted-tree.qza \ #输出的有根树
--p-n-threads 8
绘制稀释曲线
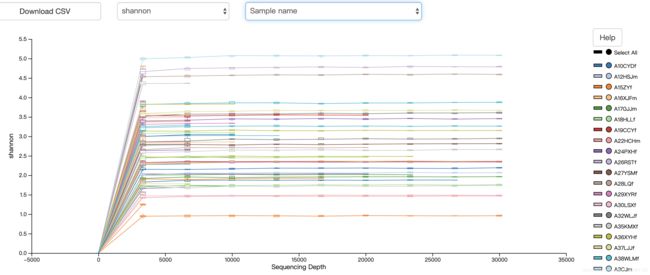
为了检查样本测序量是否足够反映真实的物种情况,需要抽样检测:原理是按照一定的梯度抽取样本reads,检测OTU数量,当OTU数量随着梯度增加不再增加时,则说明测序深度足够。
qiime diversity alpha-rarefaction \
--i-table table.qza \
--i-phylogeny rooted-tree.qza \#可选,有的话可以在计算observed_otus, shannon之外,计算faith_pd
--p-max-depth 30000 \#最大采样深度,低于该阈值的样本将被丢弃
--m-metadata-file metadata.tsv \#结果可以按照元数据信息分别显示
--o-visualization alpha-rarefaction.qzv
可选择不同的评估方法(observed_otus, shannon和faith_pd)或者查看在不同分组之间的情况。

 要注意的是系统变量LANG需要设置为
要注意的是系统变量LANG需要设置为en_US.utf-8,否则容易出现字符编码错误,报错:
![]()
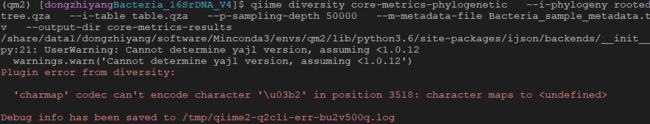
需要export LANG=en_US.utf-8,或者写到环境变量
计算物种多样性
基于feature table,通过生成一系列的系统发育和非系统发育多样性度量对物种数量和丰度二维信息进行描述,从而评估物种多样性。
qiime diversity core-metrics-phylogenetic \
--i-phylogeny rooted-tree.qza \
--i-table table.qza \
--p-sampling-depth 10000 \#为了比较序列深度不均匀的样本,样本中的最小序列数可用作采样深度,也可以稍微降低一点,可根据feature表的统计结果中Frequency per sample中minimum frequency来确定;需要注意的是,低于这个阈值的样本将会被丢弃。
--m-metadata-file ${metadata} \
--output-dir core-metrics-results
core-metrics-results目录同时包含qza和qzv数据:
- α多样性:
香农(Shannon’s)多样性指数(群落丰富度的定量度量,即包括丰富度richness和均匀度evenness两个层面)
Observed OTUs(群落丰富度的定性度量,只包括丰富度)
Faith’s系统发育多样性(包含特征之间的系统发育关系的群落丰富度的定性度量)
均匀度(或 Pielou’s均匀度;群落均匀度的度量) - β多样性
Jaccard距离(群落差异的定性度量,即只考虑种类,不考虑丰度)
Bray-Curtis距离(群落差异的定量度量)
非加权UniFrac距离(包含特征之间的系统发育关系的群落差异定性度量)
加权UniFrac距离(包含特征之间的系统发育关系的群落差异定量度量)
同样打开qzv文件可查看结果,以Bray_curties_emperor.qzv为例展示基于Bray_curties距离的PCoA:

最为常用的操作包括:
依据分组信息显示不同的颜色和形状(第二个特征),将背景设置成白色,保存为矢量图等,设置好以后如下图:
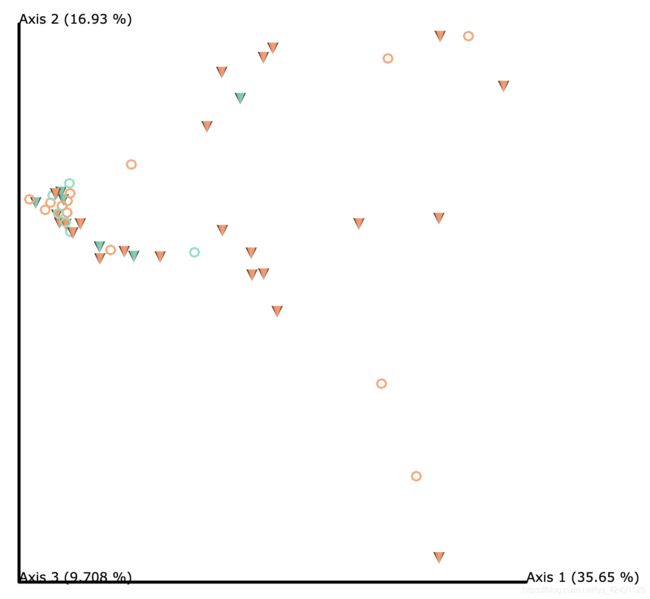
如果觉得qiime在线绘图效果不满意,可以解压相应qza文件获得画图数据,再通过R自行绘制。
物种组成分析
在获得特征表以后,我们关注的另外一个问题是每个样本究竟包含哪些物种,这些物种的相对丰度如何,这就需要进行物种注释。不同类型的扩增子数据有不同的注释数据库,这里以常见的两类数据为例:
| 类型 | D1 | D2 |
|---|---|---|
| 16S V4 | GreenGene | SILVA |
| ITS1 | UNITE | - |
qiime2有三种物种分类方法,classify-sklearn、classify-consensus-blast和classify-consensus-vsearch。sklearn是基于Naive Bayes classifier的机器学习,blast和vsearch基于序列比对,区别是前者主要基于BLAST+进行局部比对,而后者基于VSEARCH进行全局比对也更耗时。首先介绍classify-sklearn。
基于sklearn方法
- 已有训练好分类器
有过机器学习经验的应该知道,对于任何分类器模型,都需要首先基于训练集采用相应的算法提取数据特征从而构建分类器模型,建好的分类器就可以用于预测、判定新的数据。
qiime2官网已经有训练好的sklearn的分类器,只需要下载即可:sklearn分类器。
这里以Greengenes 13_8 99% OTUs为例,该分类器适用于250bp左右的16S V4区序列 (515F/806R primer pair)。
curl -sL \
"https://data.qiime2.org/2020.2/common/gg-13-8-99-515-806-nb-classifier.qza" > \
"gg-13-8-99-515-806-nb-classifier.qza" #下载分类器
conda install --override-channels -c defaults scikit-learn=0.21.2 #更新sklearn分类器包
qiime feature-classifier classify-sklearn #物种注释
--i-classifier gg-13-8-99-515-806-nb-classifier.qza \ #分类器
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza #输出物种注释信息,物种注释时间较长,建议nohup
qiime taxa barplot \ #绘制样本物种组成柱状图
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--m-metadata-file metadata.tsv \
--o-visualization taxa-bar-plots.qzv
打开taxa-bar-plots.qzv查看结果:
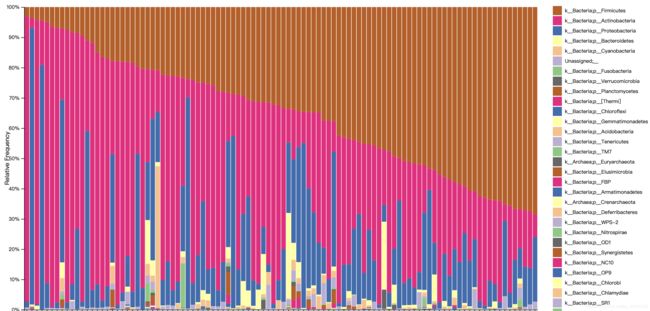 按照分组信息排序或者选择不同分类级别进行查看。
按照分组信息排序或者选择不同分类级别进行查看。
- 自行训练数据
如果没有合适的分类器,可以自行构建分类器,需要两个文件,一是OTU参考序列,而是对应的分类信息,可从等Qiime官网、Greengene和Silva等地下载。格式如下:
1.序列文件silva_97_otus.fasta
.
2.分类信息:silva_97_otus_taxonomy.txt
有了这两个文件之后就可以构建和训练分类器了。
qiime tools import \ #导入序列
--type 'FeatureData[Sequence]' \
--input-path silva_97_otus.fasta \
--output-path silva_97_otu.qza
qiime tools import \ #导入分类信息
--type 'FeatureData[Taxonomy]' \
--input-format HeaderlessTSVTaxonomyFormat \#如果没有描述信息的第一行使用此
--input-path silva_97_otus_taxonomy.txt\
--output-path silva_97_otus_taxonomy.qza
qiime feature-classifier fit-classifier-naive-bayes \#训练分类器
--i-reference-reads silva_97_otu.qza \#上一步导入的参考序列
--i-reference-taxonomy silva_97_otus_taxonomy.qza \ #上一步导入的分类信息
--o-classifier silva_97_otu_classifier.qza #训练好的分类器,耗时较长,建议nohup
有了分类器,后面的步骤与之前的步骤一样,需要注意的是sklearn的表现性能取决于训练数据的好坏,训练集越准确分类效果越好。
基于blast和vsearch方法
基于blast和vsarch方法需要refseq.qza序列和taxonomy.qza分类信息两个文件,可从以下网址(不限于)下载:
https://www.arb-silva.de/no_cache/download/archive/qiime/
https://www.arb-silva.de/download/archive/qiime
需要注意的是要下载与参考序列版本对应的物种分类文件,如果没有现成的qza文件,可从refseq.fasta序列和taxonomy.txt文件导入,方法与之前一样。
有了refseq.qza和taxonomy.qza文件就可以开始物种注释:
qiime feature-classifier classify-consensus-blast \ #blast 方法
--i-query rep-seqs.qza \
--i-reference-reads refseq.qza\
--i-reference-taxonomy taxonomy.qza \
--p-perc-identity 0.9 \ #序列相似性,默认0.8取值[0,1]
--p-query-cov 0.9 \ #覆盖率,默认0.8
--o-classification taxonomy.qza
qiime feature-classifier classify-consensus-vsearch \ #vsearch 方法
--i-query rep-seqs.qza \
--i-reference-reads refseq.qza\
--i-reference-taxonomy taxonomy.qza \
--p-perc-identity 0.9 \ #序列相似性,默认0.8取值[0,1]
--p-query-cov 0.9 \ #覆盖率,默认0.8
--p-threads 8 \#线程数
--o-classification taxonomy.qza
在物种注释完成后,即可参照之前的方法绘制物种注释柱状图。
qiime2其他常用操作
导出制表符分割的特征表
qiime2的特征表位于table.qza,需解压成feature-table.biom,再通过biom转换成制表符分割文件。
qiime tools export \ # 解压特征表,生成feature-table.biom
--input-path table.qza \
--output-path export #输出目录而不是文件
# 将biom格式转换成tsv格式便于查看
biom convert
-i feature-table.biom \
-o feature-table.tsv \
--to-tsv #转换为制表符分割文件
折叠物种分类单元
按照物种注释结果将特征表折叠成不同分类单元输出成表格:
# 默认是feature ID,需要转换成物种分类名
qiime taxa collapse \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--p-level 6 \ #分类单元
--o-collapsed-table table_level6.qza
随后可按之前的步骤导出特征表即可。
按样本筛选特征表
在物种多样性分析之后,可以看到不同样本的变异情况,有些样本可能由于批次效应与其他样本相差甚远,可以考虑去除,将待保留的样本名称保存至文件sampleid.txt,即可生成过滤的特征表,sampleid.txt格式如下:
SampleID
B31ZRf
B43LJDm
A5CMCm
A30LSXf
MCB.M.C5
qiime feature-table filter-samples \
--i-table table.qza \
--m-metadata-file sampleid.txt \#保留样本名称
--o-filtered-table filter_by_id_table.qza
至此,扩增子分析最常见的几张图就完成了。后续将介绍PICRUSt、LEfSe和Co-Occurrence网络分析。
参考
https://docs.qiime2.org/