2020.08.18【转载】丨叶绿体基因组二代测序组装经验分享
叶绿体基因组二代测序组装(个人经验分享)
前段时间,有老师咨询我关于叶绿体基因组组装的问题,虽然本人不才,但也很热心地帮了个忙。虽说中间出了一些小意外,唉唉算了还是不提了。在这里顺便就个人常用的叶绿体基因组组装思路和方法(基于二代测序),给大家作个分享。
叶绿体基因组本身不大(平均不到200kb),所以使用二代测序,在高深度测序模式下,配合一个有效的参考基因组,理论上足以组装出一条完整的环状序列出来(10个里面9个可以吧)。当然,只单纯地通过组装软件自动拼接基本上是不可能实现的(主要是IR区的问题,组装软件无法有效识别这两段IR序列),中间还有些小技巧。
话不多说了,上正文。不过因为本文是经验分享,不是软件使用详解,所以很多内容建立在已知的基础上,不是很适合纯新手阅读(至少要了解过基因组组装、熟悉linux使用)。
叶绿体基因组的二代测序组装过程(个人常用经验)
以下是本人常用的叶绿体二代组装过程,期间结合使用了多种工具,不乏手动操作的环节,过程或许比较繁琐。但综合考虑的情况比较多,所以结果一般都是很理想的。可能大神们有更有效的、更方便快捷组装思路,还希望分享学习哈。
以下涉及的软件,对于它们的下载安装、怎么使用、结果解释、以及可能存在的报错问题等,不作描述;以及一些常规概念,默认大家已知了。
此外,本文的方法建立在存在有效的近缘物种叶绿体参考基因组的基础上,算是一种“有参组装”策略。若是你的目标叶绿体,未能找到一个可靠的参考基因组,请忽略本文吧。要问无参考基因组的情况应该怎样做,测序公司一般建议你上三代测序;至于我,我不知道……
0、准备文件
测序数据clean reads
首先是测序数据,我们直接从fastq格式的测序数据clean data(clean reads)开始。如下所示,通常情况下,为了节省空间会将fastq压缩为gz格式存储。
![]()
参考叶绿体基因组
组装期间需要近缘物种的叶绿体基因组做参考,主要用来解决以下两种问题:
(1)测序数据中存在大量的核基因组,那么首先就是要尽可能去除核基因组对叶绿体基因组组装的影响。使用参考叶绿体基因组,在测序clean data中“挑选”出叶绿体基因组的测序reads,后续使用这些挑选后的reads作组装,快速、高效、准确。
(2)基于二代测序数据,基本上是无法仅通过拼接软件一次将叶绿体基因组自动拼接成环的,通常会得到多条contigs/scaffolds序列。后续还需参照参考基因组,对这些contigs/scaffolds序列定位和定向。
假设我们的叶绿体来源于Saccharina属的某物种,那么首先需要在NCBI或EMBL等数据库中,寻找一些其近缘物种的叶绿体基因组。
可通过“Saccharina chloroplast”、“Saccharina plastid”等关键词查询同属物种的叶绿体基因组。同属的没有,同科的也凑合吧,再远一些,真的不怎么好使了。尽管植物叶绿体比较保守,但亲缘关系较远的话,还是会有较大的差异,差异过大时非常影响二代组装质量。此外,下载时尽可能多下几个参考基因组作备选。
我们下载了多条序列,合并在一起,命名为“ref.fasta”。
##0、准备
#新建路径
mkdir -p ./chloroplast/0-data ./chloroplast/0-refer
#将测序数据 clean data 移至(或软连接至) 0-data
#参考基因组存放至 0-refer
#多个参考基因组 ref1.fasta、ref2.fasta、ref3.fasta 等,cat 到一起使用:
#cat ref1.fasta ref2.fasta ... > ref.fasta
1、“挑选”叶绿体基因组测序reads
第1步,需要尽可能地将clean data中的核基因组测序reads去除,保留叶绿体测序reads,便于后续组装的精度与效率。
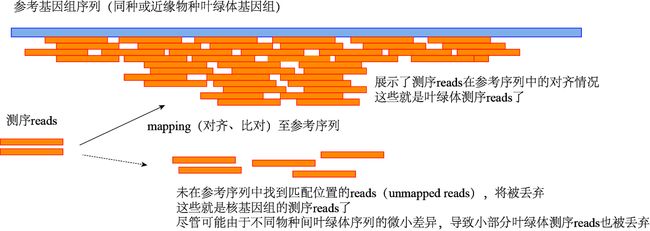
这一步我们借助参考基因组来完成。常用方法,将测序clean reads对齐(mapping)至参考叶绿体基因组中,保留能在参考基因组中对齐的测序reads。这样,属于叶绿体基因组的测序数据就筛选出来了。那些未能对齐至参考基因组中的测序reads,可以认作是核基因组,去除即可。
当然,不可否认,由于不同物种的叶绿体之间还是存在微小差异,因此这种方法可能会错误地将很少一部分叶绿体基因组测序reads去除(测序物种叶绿体上的某些小段序列,可能在参考基因组中没有,因此会被过滤掉),导致组装出现gap。不过影响不大,后面会提到怎样填补gap。
能够实现该功能的工具有很多。
例如,使用BWA作测序reads与参考基因组的对齐,获得比对结果bam文件;使用SAMtools工具,过滤掉其中未匹配的测序reads,并对bam文件中保留的reads作排序(按测序reads的id排序),最后在bam文件中将这些保留的reads提取出得到筛选后的fastq文件。
##1、挑选 reads 序列
#首先给参考基因组建立 BWA 索引
cd 0-ref
bwa index ref.fasta
#使用 BWA 将测序 clean reads 对齐至参考基因组获取 bam 文件,并使用 SAMtools 将能够对齐的 reads 挑选出得到筛选后的 fastq 文件
注:以下 samtools fastq 功能,必须源码编译 SAMtools 才具有,conda 安装的不具备该功能。
cd .. && mkdir 1-mapping && cd 1-mapping
bwa mem -t 4 ../0-ref/ref.fasta ../0-data/test_chloroplast_cleandata_R1.fastq.gz ../0-data/test_chloroplast_cleandata_R2.fastq.gz | samtools view -@ 4 -bS -F 12 | samtools sort -@ 4 -n | samtools fastq -@ 4 -1 bwa_R1.fastq -2 bwa_R2.fastq -本示例,过滤reads后得到的新fastq文件命名为“bwa_R1.fastq、bwa_R2.fastq”,用于后续的组装。
2、基因组的二代初步组装
第2步,我们就可以直接使用上一步挑选出来的测序reads作组装了。嗯嗯,组装过程就不用再画个图示了吧。
基因组二代组装工具挺多的,比如SOAPdenovo、ABySS、SPAdes、A5-miseq等。这一步多尝试几个拼接软件(至少两个吧,只用一个可能不太够用,原因后面会提到),分别得到不同的拼接结果。
例如分别使用SPAdes和A5-miseq作组装。对于其它组装工具,我就不作更多演示了,总之大家根据实际情况选择吧。
##2、基因组组装,使用筛选后的 fastq 文件进行基因组的二代组装
cd .. && mkdir 2-assembly && cd 2-assembly
#使用 SPAdes
spades.py -1 ../1-mapping/bwa_R1.fastq -2 ../1-mapping/bwa_R2.fastq -o ./spades -t 4 --cov-cutoff auto --careful
#使用 A5-miseq
a5_pipeline.pl --threads=4 ../1-mapping/bwa_R1.fastq ../1-mapping/bwa_R2.fastq a5miseq
*组合1、2步的工具
一些组装工具,如ARC(Assembly by Reduced Complexity)、NOVOPlasty等,集合了测序reads与参考基因组的比对、提取,以及组装过程,提供测序clean data与参考基因组即可自动装配。此外部分工具还可以自动基于参考基因组实现组装序列的定位定向(见下文)过程。听起来倒是蛮方便的,但是本人并没有使用过不知实际效果咋样,大家有兴趣可以测试下。
3、contigs/scaffolds的定位和定向
好了从这步开始需要结合手动的过程了。
经过初步拼接后,我们获得了几个组装结果fasta文件。这些fasta文件中通常存在多条contigs/scaffolds序列(仅凭软件自动组装得到一整条序列,几乎不太可能),下一步就需要确定这些contigs/scaffolds序列在基因组中的相对位置和方向(定位和定向),以继续往完整的环状叶绿体基因组序列搭建。
这一步也需借助参考基因组来完成。将组装得到的那些contigs/scaffolds序列与参考基因组对齐,确定位置和方向关系。

同样地,能够实现该功能的工具有很多,可视化的工具如geneious,命令行工具如MUMmer,等等。
本示例这里选择了共线性分析工具MUMmer来完成。先以某个组装结果fasta文件为例。
##3、共线性分析
#MUMmer 共线性分析定位 contigs/scaffolds 的顺序
#以上述 SPAdes 组装结果 fasta 文件“scaffolds.fasta”为例
cd .. && mkdir 3-mummer && cd 3-mummer
nucmer --mum -p chloroplast ../0-ref/ref1.fasta ../2-assembly/spades/scaffolds.fasta
delta-filter -m chloroplast.delta > chloroplast.filter
show-coords -T -r -l chloroplast.filter > chloroplast.1coords
mummerplot --postscript -p chloroplast chloroplast.delta
ps2pdf chloroplast.ps chloroplast.pdf
#若你的 ref.fasta 是多个参考基因组 fasta 文件中的序列 cat 到一起的,请不要直接使用这个混合的
#请在原始参考基因组文件中找一个最近缘物种的 fasta 文件使用,比方说这里的 ref1.fasta对于共线性分析结果,可以直接查看文本结果文件“chloroplast.1coords”中的内容,记录了组装scaffolds序列和参考基因组序列的共线性匹配详细信息。或者更直观的,查看最后生成的共线性结果图,如下所示这样。

据此,我们可以确定组装结果中scaffolds序列的定位定向结果大致是下图中这样的。下方为参考叶绿体基因组序列,上方为组装所得scaffolds序列,其中共涉及到4条重要的scaffolds序列,红色表示正向,蓝色表示反向(所谓“反向”,意思就是后面我们要取它的反向互补序列)。

到这一步,我们基本确定了叶绿体基因组的轮廓了。
4、基因组环化
根据上一步的contigs/scaffolds定位和定向结果,把符合条件的序列挑选出来,之后查找这些contigs/scaffolds序列之间是否有重叠(overlap)区域,并根据这些overlap区域连接序列。
对于contigs/scaffolds序列的挑选,可使用SAMtools等工具,或者自写脚本提取,就算手动打开fasta文件选择序列也不麻烦。该反向互补的注意反向互补。之后可继续结合共线性分析、BLAST比对等工具来确定前后contigs/scaffolds序列之间是否有高可信度的一致区域。就算你想偷个懒,手动根据序列起始/或末尾处的碱基文本在fasta文件中搜索匹配一致的序列也行。最后确定这些重叠区域都位于contigs/scaffolds序列中的哪些位置,对序列裁剪、连接。
具体操作过程不再细说了,上段文字说明挺易懂的吧。

可知,要是所有contigs/scaffolds序列之间全部存在重叠区域,即可连接成环。运气好的话,此时的序列既无gap,又成环了,这个叶绿体基因组的组装基本上就算完成了。此时你可以跳到第6步的内容。
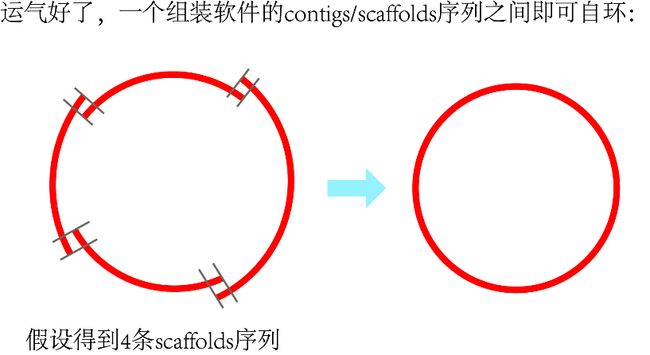
当然实际情况中,运气不可能总是这么的好,总有无法连接起来的gap。那么,第5步的内容会有所帮助。
5、填补gap
我看网上的关于叶绿体基因组组装的教程,基本上都只介绍了reads过滤、二代组装以及序列定向和环化这些环节,并没有细说应该怎样去补齐基因组中的gap。好吧,若有gap出现,补gap肯定是最糟心的一步了。对于怎样将叶绿体基因组序列中的gap尽可能填补,以下提到的方法主要基于个人经验。经验尚浅,大家若是有更好的方法,还请分享哈。
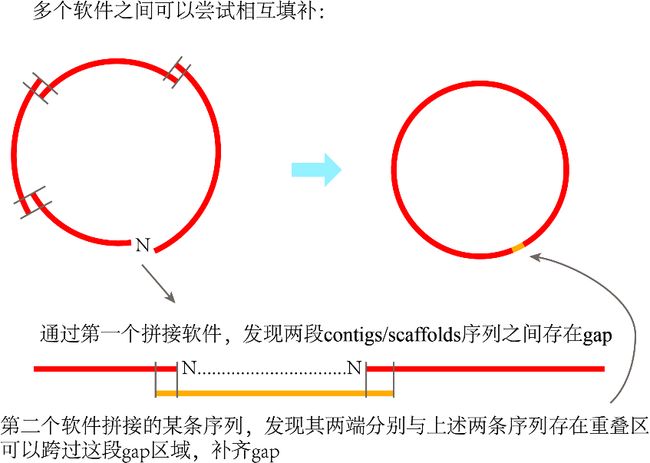
多个组装软件的结果相互结合
在第2步组装时,建议使用多个软件作拼接。我们可知,不同的软件拼接效果肯定有所差异。
首先,运气好的话,某个软件的结果,所有contigs/scaffolds序列之间全部存在重叠区域,可以自己成环,多尝试几个软件说不定就能碰到这个运气。
其次,对于出现gap的位置,可以结合多个软件的拼接序列,相互搭建,看不同软件所得序列之间是否能够相互填补空缺。一个软件没拼出来的区域,另一个软件拼出来了,很常见。环化方法参考第4步。
结合从头组装的结果
在第1步,组装之前,为了去除核基因组的影响,以及提升组装效率,我们首先根据参考基因组过滤了核基因组reads。同时提到,这种方法可能会因不同物种叶绿体基因组间的微小差异,而错误地将很少一部分叶绿体基因组测序reads去除,这些reads若是集中在基因组的某些特定区域,则会导致组装时这些区域缺失,出现gap。
此时不妨再拿原始的所有clean reads做个从头组装,然后再通过共线性分析等方法,查看是否有contigs/scaffolds序列可以跨越先前结果中的gap区,以连接空缺。环化方法参考第4步。

*CAP3工具
对于上述两种方法,若是觉得手动一个个寻找序列间的重叠区很吃力的话,其实也有工具能够帮助自动实现。例如CAP3,早期它主要用在sanger序列的拼接中,其实对于像叶绿体、线粒体、小型质粒等这样的超小型基因组,我们也可以借助它来辅助基因组环化。
不过CAP3的严谨性不高,有小概率出现错误拼接的位点,所以这里我给这个工具打了星号标记。虽然说大部分情况还是很靠谱的。你可以在后续将测序clean reads比对到组装后的基因组上,通过IGV、Tablet等基因组可视化工具,检查基因组中是否有不严谨的拼接位点。
#假设这里有多种组装软件的拼接结果:spades.fasta、a5.fasta、soap.fasta
#我们可尝试借助 cap3 识别这些序列之间的 overlap 区并相互延伸
cat spades.fasta a5.fasta soap.fasta > all.fasta
cap3 all.fasta > cap3.log事实上,像这类的拼接小片段序列的工具还有很多,大家也可自行搜索,试下效果。
gap填补工具
事实上,目前有很多工具能够直接使用测序reads,填补序列中N碱基空缺。因此对于gap区域,我们可以首先拿N碱基连起来(具体多少数量的N,可以参照参考基因组来确定),然后再设法补洞。
例如SOAPdenovo套件中的GapCloser等,可以试一下,还是能填补不少的N空缺的。
#GapCloser 补洞(config,SOAPdenovo library文件)
GapCloser -b config -a input.fasta -o output.fasta
手动延伸
这个是最无奈方法了,实在没招时考虑下吧,可能会非常繁琐。就是使用测序clean reads手动一点点延伸。
首先在gap两端的序列中,在5’端序列的末尾截上150bo左右,3’端序列的起始位置同样截上150bo左右。之后将clean reads与这两小段序列对齐,根据能比对上的reads序列碱基组成,手动对gap两端的序列进行延伸。根据测序策略的不同,一次操作能同时在两端延伸大约100bp(PE150)或200bp(PE250、PE300)不等。如此循环,直到reads能把两端接起来。
如果使用这种方法发现了测序断点,即在某段区域不再存在reads支持,两端的序列无法再通过reads继续延伸,但gap仍未连接起来时,则表明基因组中确实存在了测序未测到的区域。不过这种真的是小概率事件了,碰到真的没招了,至于为什么出现这种现象,根据我的经验,这些区域主要是重复序列结构(而且是高AT那种,我们知道二代测序普遍存在GC偏好性,导致位置很难扩增)。或者,特殊的碱基组成导致测序时单链序列产生了二级结构?其实就算再重新测序,还是不一定有把握把这些位置测出来……要问我怎么解决,我也不知道啊……(本文开篇提到的帮那位老师组装的叶绿体,小意外就是测序数据就出现这种问题了,唉唉个人已经尽力了![]() )
)
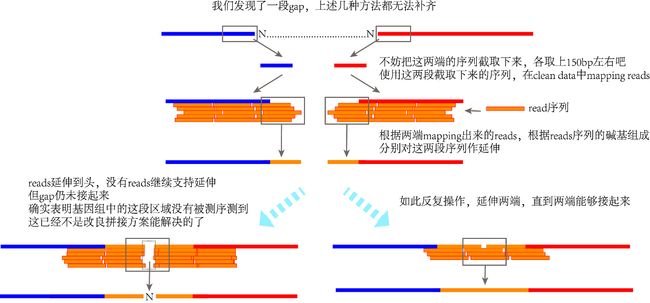
例如,截取两端序列,新建一个fasta文件,结合BWA、SAMtools将clean reads对齐到这两端序列上,只保留能对齐的reads,得到SAM文件。这里默认大家已知SAM文件的结构了。可知这个SAM文件的体积不大,直接拿文本编辑器打开它即可。查看匹配reads的碱基组成,根据“悬挂序列”,手动在原组装结果中延伸gap两端的序列,缩短gap。如此循环。方法很好理解,结果也很有效,要是数据质量够好(不存在没测到的基因组区域),无论初始拼接的基因组再差,最后都一定能成环,就是非常麻烦了。
bwa index cut.fasta
bwa mem -t 4 cut.fasta test_chloroplast_cleandata_R1.fastq test_chloroplast_cleandata_R2.fastq | samtools view -@ 4 -bS -F 12 | samtools sort -@ 4 > bwa.bam
samtools view bwa.bam > bwa.sam
*一代测序
若有把握gap在1kb之内,可以尝试设计引物扩增这段区域,使用一代测序得到它。不过这个已经不属于分析方法的范畴了,简单一提带过。
6、碱基校正
环化完成后,完整的叶绿体基因组序列拼接好了,后续再加个碱基校正的过程就OK啦。
Illumina的二代测序错误率约1%,也就是平均每100个碱基中会出现一个错误碱基,可能表现为“碱基的突变”,也可能表现为“碱基的插入/缺失”。在组装过程中,这种错误可能会被引入,使得基因组中的某些位点不太可靠(如,出现模糊碱基,或者极个别的N碱基)。
我们可以将测序clean reads重新与我们的组装结果基因组序列对齐,根据高深度的覆盖reads去识别、校正模糊的位点。
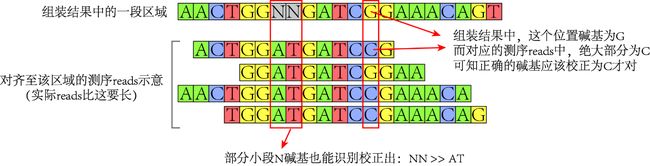
例如,结合使用BWA、SAMtools实现对齐过程,使用Pilon软件对基因组进行校正。
#pilon,final.fasta 是最终环化后的结果
mkdir 6-pilon && cd 6-pilon
bwa index final.fasta
bwa mem -t 4 final.fasta ../0-data/test_chloroplast_cleandata_R1.fastq ../0-data/test_chloroplast_cleandata_R2.fastq | samtools view -@ 4 -bS -F 12 | samtools sort -@ 4 > bwa.sort.bam
samtools index bwa.sort.bam
pilon --genome final.fasta --frags bwa.sort.bam --output final.pilon1 --changes --threads 4对于本示例,Pilon校正后,得到两个文件,“final.pilon1.fasta”和“final.pilon1.changes”。若是changes文件大小为0(空文件),则其对应的fasta文件可作为最终结果;若不为0,则还需再基于新得到的fasta文件重新执行校正;以此类推,直到changes文件为0为止。有时候可能由于测序数据本身质量较差的问题,多次循环后未见0大小的changes文件,则你可以终止重复校正过程,从已得到的所有changes文件中找一个最小的,其对应的fasta文件可作为最终结果。
事实上,由于Pilon也可以识别小段的N碱基序列,因此除了用于碱基校正外,或许也能用于填补小部分gap。
7、最终检查
不要高兴太早,最后一定不要忘了好好检查。
我们可以将原始的clean reads比对到最终的叶绿体基因组上,得到bam文件,然后使用基因组浏览器(如IGV、Tablet等)打开bam文件以及基因组fasta文件,仔细查看测序reads在基因组序列中的覆盖情况,判断:
(1)reads覆盖是否均一,基因组中哪些区域reads覆盖深度较少,低深度的reads是否能有效支持这一段序列的拼接。
(2)是否存在疑似错拼的位点,比方说某个位置两端的reads出现很大“断层”。
(3)是否还有个别位置的碱基疑似由测序错误导致的类似于“SNP”、“InDel”情形。
(4)对于重复序列较多的区域,reads是否能够有效跨越。
8、调整起点
关于环状基因组序列的起点位置调整,可以参照参考基因组的起点位置来调,或者根据基因组注释结果来。切记首尾一定不要出现“断开”的基因。
9、关于叶绿体注释
叶绿体注释比拼接还折腾,大家慢慢玩,我先撤了……
我在前文分享过两个叶绿体基因组注释工具,GeSeq和PGA,希望有所帮助。听一老师提到使用geneious进行叶绿体注释挺好用的,但我还没试过,大家可以试下。更多其它的方法,还请自行研究了。