药物基因组学_个体化实验分析_实验报告
文章目录
-
- 一、实验目的
-
- 1、(学会)收集特定癌种特定药物的包含药物响应与否相关的基因表达谱数据(TCGA/GEO)
- 2、(掌握)根据响应信息将样本划分为耐药或者敏感两类
- 3、(掌握)构建分类器
- 4、(学会)利用构建的分类器实现个体化耐药预测
- 二、实验流程
-
- (1)搜集数据【数据使用的是平时研究的癌型】
- (2)根据响应信息将样本划分为耐药或者敏感两类
- (3)构建分类器【以下使用的分类器是weka软件中所构建的】
-
- ①筛选差异基因
- ②构建放入weka软件的训练集和验证集文件
- ③接下来就可以放入weka进行分析
-
- \1. 随机森林
- \2. svm支持向量机
- (4)利用构建的分类器实现个体化耐药预测
-
-
- \1. 随机森林
- \2. svm支持向量机
-
- 三、讨论
一、实验目的
1、(学会)收集特定癌种特定药物的包含药物响应与否相关的基因表达谱数据(TCGA/GEO)
2、(掌握)根据响应信息将样本划分为耐药或者敏感两类
3、(掌握)构建分类器
4、(学会)利用构建的分类器实现个体化耐药预测
二、实验流程
(1)搜集数据【数据使用的是平时研究的癌型】
从GEO网站上搜索 Colorecal Cancer FOLFOX【结直肠癌药物治疗联合方案】
在series_matrix文件中查找是否有药物相应的数据,如下图。
![]()
![]()
根据自己感兴趣的方向所选择的数据集GSE19860。同时还下载了GSE3964(5-FU),GSE72970(FOLFOX),但是由于在做T检验筛选差异基因的时候,后面两套数据根据药物是否敏感信息进行分类得到的t检验结果并不显著,因此,最后选择了GSE19860进行以下的个体化耐药分析实验。
(2)根据响应信息将样本划分为耐药或者敏感两类
![]()
在这套数据中,将FL_Responder定义为敏感标签为0,FL_Non_Responder定义为耐药标签为1。
(3)构建分类器【以下使用的分类器是weka软件中所构建的】
①筛选差异基因
代码:
setwd("E:\\...\\experiment")
rm(list=ls())
\######筛选差异基因
exp<-read.table("GSE19860_series_matrix.txt",header=T,sep="\t",stringsAsFactors = F)
\####补充缺失值
library(DMwR)
exp_new<-knnImputation(exp[,-1], k = 15, scale = T, meth = "weighAvg", distData = NULL)
\#exp<-exp[-which(is.na(exp[,1])),] ####观察到有缺失值,因此采用k均值补缺失值
\#length(which(is.na(exp[,1])))
labell<-read.csv("label.txt",sep="\t",header=F,stringsAsFactors = F)
geneid<-exp[,1] #####响应response敏感为0,no response不响应耐药为1
exp_normal=exp_new[,which(labell[1,]==0)]
exp_cancer=exp_new[,which(labell[1,]==1)]
label<-c(rep(0,dim(exp_normal)[2]),rep(1,dim(exp_cancer)[2]))
\##rep()表示重复,重复1这个标签exp_normal的列数([2]表示列,[1]表示行)
exprs<-cbind(exp_normal,exp_cancer)
exp_case=exprs[,which(label==1)];
exp_control=exprs[,which(label==0)];
t_result=matrix(,length(geneid),4)
for(i in 1:nrow(exp)){
T_test=t.test(exp_case[i,],exp_control[i,],alternative="two.sided",paired=FALSE)
t_result[i,1:3]=c(geneid[i],T_test$statistic,T_test$p.value);
}
t_result[,4]=p.adjust(t_result[,3],method="BH");
t_final<-t_result[t_result[,3]<0.05,1:4] ###观察到fdr<0.05的情况下,无结果,因此采用p<0.05进行筛选差异
②构建放入weka软件的训练集和验证集文件
#####构建放入weka的文件
\####对于筛选出的差异的探针,将这些探针对应的在样本中的表达谱提取出来
loc<-match(t_final[,1],exp[,1])
new<-exp[loc,]
new<-t(new) ####将数据设置为行为样本,列为基因
colnames(new)<-new[1,]
new<-new[-1,]
label_new<-as.data.frame(t(labell))
all_differ<-cbind(new,label_new)
\####训练集和测试集数据
traindata<-all_differ[c(1:20),]
testdata<-all_differ[c(21:40),]
\###选取20个样本作为训练集,20个样本作为测试集。
write.csv(traindata,"traindata.csv")
write.csv(testdata,"testdata.csv")
#####接下来是在excel表格中进行的操作
训练集和测试集的数据最后一列都是label的标签,其中测试机的label要都换成英文的?,如下图。
####放入weka将.csv的文件保存为.arff文件
步骤:Explorer -> open file [打开所要的训练集和测试集文件] ->save[保存为.arrf文件格式]
####对.arrf文件在notepad+进行操作

将label后面的numeric都换成英文的{0,1},这一步很重要,不然后续会报错。
③接下来就可以放入weka进行分析
以下分析采取20个样本作为训练集,20个样本作为测试集,效果是与其他不同样本比例划分所得到的结果相比较后是较好的了,因此采用1:1构建训练集和测试集。
注:因为采取训练集个数大于测试集个数,会导致分类器的分类结果过度拟合,而导致分类准确率下降,并且准确率都在50%。
\1. 随机森林

在cmd(命令提示符)输入如下代码:
java -Xmx1024m -classpath .;D:\Weka-3-6\weka.jar weka.classifiers.trees.RandomForest -t C:\Users\11833\Desktop\class_data\traindata.csv.arff -d colonRandomForest.model > colonRandomForest.out
![]()
以下操作也如上图进行输入,建议将weka装在D盘,路径都为英文,防止出错。
\2. svm支持向量机
在cmd(命令提示符)输入如下代码:
svm模型的构建
java -Xmx1024m -classpath .;D:\Weka-3-6\weka.jar;D:\Weka-3-6\libsvm\java\libsvm.jar weka.classifiers.functions.LibSVM -t C:\Users\11833\Desktop\class_data\traindata.csv.arff -d colonLibSVM.model > colonLibSVM.out
(4)利用构建的分类器实现个体化耐药预测
根据前面的操作,我们会得到相应的分类器模型,接下来就是对测试结果进行验证。
\1. 随机森林
代码:
java -Xmx1024m -classpath .;D:\Weka-3-6\weka.jar weka.classifiers.trees.RandomForest -l colonRandomForest.model -T C:\Users\11833\Desktop\class_data\testdata.csv.arff -p 0 > PredictionResultRandomForest.txt
%%%%这个语句是用构建的模型来预测验证集-l是载入保存的模型,-T是验证集 -p 0这个参数是给出每个样本分类的标签,和分为该类的可能性
%%%%验证集的arff文件和训练集不一样,他的类标签相当于是未知的,在类标签那列就用?表示,但是属性那里要注意,你这里要和训练集一样
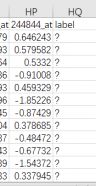
结果:

该数据是选择了训练集30个样本,测试集10个样本,根据比较,该分类器准确率只有50%。
\2. svm支持向量机
代码:
svm验证集的验证结果
java -Xmx1024m -classpath .;D:\Weka-3-6\weka.jar;D:\Weka-3-6\libsvm\java\libsvm.jar weka.classifiers.functions.LibSVM -l colonLibSVM.model -T C:\Users\11833\Desktop\class_data\testdata.csv.arff -p 0 > PredictionResultSVM.txt
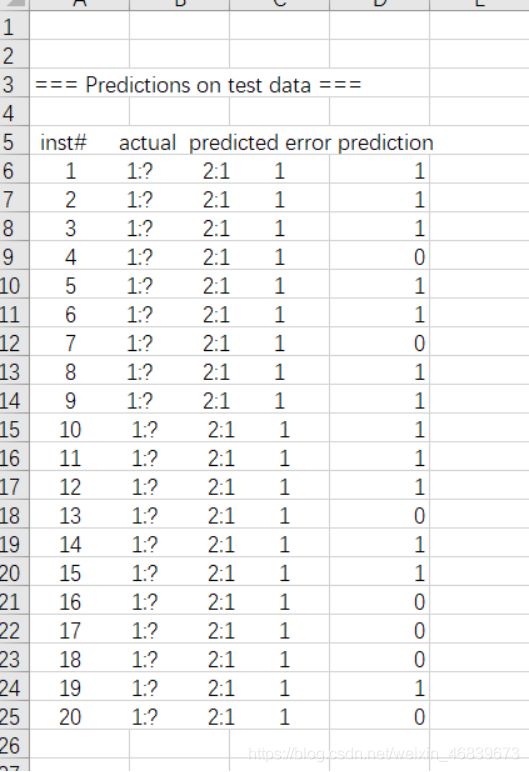
其中分类正确的共有13个,分类错误的有7个,该分类器的正确率为65%。
三、讨论
下载的三套数据都没有得出较为差异的结果,可能是因为数据的分类未能很好将差异的基因筛选出来,且我同时注意到大概只剩下1000多个探针没有对应多个基因ID,而其他探针大多都比对到了多个探针,因此仍然对数据结果有所保留,也可能因此导致了分类器不够robust,所以分类效果不够准确。

欢迎关注我的微信公众号呀~