
- 1. 摘要
- 2. 引言
- 3. 材料与方法
- 4. 结果与讨论
- 5. 参考文献
1. 摘要
草地贪夜蛾(Spodoptera frugiperda)原产于美洲,具有长距离迁飞特性,该虫于1988年入侵欧洲,2016年入侵非洲,并引起巨大粮食损失,2018年入侵亚洲,2019年从缅甸传入中国,并在短时间扩散至全国10多个省[1]。截止到这篇文章[2],目前已经有七篇关于草贪的基因组文章,包括这篇文章在内的四篇已发表文章以及三篇在预印本网站bioRxiv上的文章。
作者从浙江省采集到的单个雌蛹(ZJ-version)作为测序个体,进行PacBio和Hi-C测序。组装的基因大小为486Mb,contigN50为1.13Mb,scaffoldN50:16.3Mb。通过Hi-C构建scaffold形成31条染色体和一部分W染色体(ZW型),性染色体的鉴定是通过对一对雌雄蛹进行重测序得到。基因组的重复度为28%,蛋白编码基因为22,623个。通过比较基因组分析,解毒酶相关,化学感受相关,营养代谢,转运蛋白相关的基因家族出现明显的基因家族扩增现象,这与其多食性以及抗药性等性状相关联。
2. 引言
high fecundity, long adult life span, high spawning and strong ability to fly.
two haplotypes, the “rice strain” (R strain) preferring to feed on rice and grasses and “corn strain” (C strain) preferring to feed on corn and sorghum. 这两种单倍体型有不同的性信息素组成,对Bt蛋白转基因植物或杀虫剂的敏感性不同以及繁殖行为不同。
区分这两种类型:the sequence of mitochondrial Cytochrome Oxidase Subunit I (COI) and strain-specific sites in the fourth exon of the Z-chromosome-linked gene Triosephosphate isomerase (Tpi).
3. 材料与方法
3.1 基因组测序和从头拼接
One SMRT cell was run for genome sequencing. PacBio sub-reads were initially cleaned using Canu(https://github.com/marbl/canu) for sequence error correction.
The PacBio corrected reads were assembled by SMARTdenovo (https://github.com/ruanjue/smartdenovo).
The redundans pipeline (https://github.com/lpryszcz/redundans) was used to remove the redundant contigs from the initial de novo assembly genome with the parameters “--identity 0.5, --overlap 0.75”.
3.2 Hi-C 文库制备
Sixth instar larva; Illumina HiSeq platform with 2×150 bp reads.[^8]
3.3 Hi-C构建scaffold
HiC-Pro pipeline (https://github.com/nservant/HiC-Pro) was used to identify valid read pairs.
each read in the pair is mapped independently and, where ligation sites are detected by exact matching, the 3’ sequence is trimmed from the read and the 5’ portion remapped.
The sequence alignments were made using Bowtie2 with the parameters “--very-sensitive -L 20 --score-min L,-0.6,-0.2 --end-to-end --reorder --rg-id BMG --phred33-quals -p 5”. The processed mappings were then merged into a single alignment file with valid interaction pairs expected to involve two different restriction fragments.
Then the valid interaction pairs were used to build the interaction matrices and we scaled up the primary genome assembly contigs into chromosome-scale scaffolds (hereafter pseudo-chromosomes) with LACHESIS (https://github.com/shendurelab/LACHESIS).
To access the accuracy of the scaled-up genome assembly, we cut the pseudo-chromosomes predicted by LACHESIS into bins with 100 kb lengths. Then we constructed a heatmap based on the interaction signals that were revealed by valid mapped read pairs between bins. The matrix was produced by HiC-Pro and then visualized as a heatmap to show the diagonal patches of strong linkages.
3.4 转录组测序和分析
Larva (from first instar to six instar), female pupa, female adult and male adult of fall armyworm were sequenced using the Illumina HiSeq 2000 platform with paired-end libraries(Three biological replicates).
Low-quality bases in the RNA-Seq raw reads were first filtered using Trimmomatic. Then, the clean reads were mapped to the genome assembly using Hisat2 and StringTie to obtain putative transcripts.
To determine gene expression levels, the RNA-Seq clean reads were mapped to the genome assembly using Bowtie2, and transcript abundances were estimated by RSEM.
3.5 基因组组装质量评估
BUSCO v3.0(Benchmarking Universal Single-Copy Orthologs) software to scan 1,658 universal single-copy orthologous genes selected from insecta_db 9 data sets in genome assembly with default parameters.
3.6 基因组注释
Repeat sequences and transposable elements (TEs)
- For de novo predictions, RepeatModeler was used to construct a de novo repeat library with default parameters. For homology-based predictions, RepeatMasker was used with Repbase library.
**The protein coding genes **
By integrating the evidence of de novo, homology-based and RNA-Seq-based annotations
Augustus and SNAP were used to generate the de novo annotation with internal gene models.
Exonerate and GenomeThreader were used to align the proteins obtained from NCBI invertebrate RefSeq to the genome assembly with default parameters.
The transcripts of the fall armyworm were obtained by Hisat2 and StringTie pipeline with default parameters.
We next integrated these three types of evidences with different weights (the weight for de novo annotation is “1”, for homology-based annotation is “5”, for RNA-Seq-based annotation is “10” ) for each by EVidenceModeler (EVM) to obtain the official gene set (OGS).
Gene Ontology (GO) analysis was carried out using the software Blast2GO. We further mapped these genes to data from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database using the BlastKOALA online service.
3.7 进化分析
Proteins sequences of 22 insect species were clustered using the OrthoMCL pipeline with default parameters.
328 single-copy genes were obtained from OrthoMCL results and were used for phylogeny reconstruction.
The protein sequences of each gene family were independently aligned by MAFFT. Then, trimAl was used to clean each alignment and extract the conserved block.
We concatenated all single-copy genes to create one super gene for each species. We used ModelFinder to select the best model. IQ-Tree was used to construct the phylogenetic tree using the LG+F+I+G4 model and 1,000 bootstrap replicates.
To estimate the divergence time of the fall armyworm, we applied three calibration points based on fossil records in Paleobiology Database (www.paleobiology.org): 1) stem Trichoptera (Phryganea solitaria) at 311.45–314.6 mya; 2) stem Lepidoptera (fossil unnamed) at 201.3–208.5 mya; and 3) stem Noctuoidea (Noctuites incertissima) at 28.1–33.9 mya.
The divergence time was estimated by using MCMCtree in PAML with the topology of these insects we built above. The tree was visualized using FigTree.
3.8 全基因组共线性
Whole-genome synteny between S. frugiperda, S. litura, and B. mori were estimated using Satsuma, a package of SPINES with default parameters (https://www.broadinstitute.org/genome-sequencing-and-analysis/spines).
Synteny blocks were plotted across chromosomes using Circos.
3.9 基因家族的收缩和扩张
We used CAFÉ to perform a gene family expansion and contraction analysis. The protein sequences from twenty-two insects were aligned to the TreeFam v9 database to obtain the TreeFam ID for each protein.
The TreeFam v9 results and a tree with estimated divergence time were used as inputs of CAFÉ. We used a criterion of P < 0.05 for significantly changed gene families.
3.10 基因家族分析
P450 gene family
The P450 gene family, we first downloaded reference protein sequences of Lepidoptera P450s from NCBI GenBank and manually confirmed these sequences to obtain a clean reference sequences for Lepidoptera P450s. TBLASTN was used to search P450 candidate sequences in the fall armyworm genome assembly (E-value < 1E-5).
Genewise and Exonerate were used to define the gene structure. We also confirmed the P450 candidate sequences using HMMER against sequences from the Pfam database (Pfam domain PF00067, E-value < 1E-5).
The fall armyworm P450 sequences were compared to P450 genes of S. litura and B. mori by phylogenetic studies for name assignment. RSEM was used for gene expression level (FPKM) calculation.
Gustatory receptor (GR) gene family
The gustatory receptor (GR) gene family, we searched GR candidate sequences in the fall armyworm genome assembly using TBLASTN (E-value < 1E-5) with a set of GR reference sequences obtained from NCBI GenBank.
Genewise and Exonerate were used to define the gene structure.
For GR subfamily annotation, we compared the fall armyworm GR sequences with GRs from S. litura and B. mori by phylogenetic studies. RSEM was used for GR gene expression level (FPKM) calculation.
For other gene families
Including glutathione-S-transferases (GSTs), carboxylesterases (COEs), ATP-binding cassette transporters (ABC transporters), olfactory receptors (ORs), ionotropic receptors (IRs), odorant-binding proteins (OBPs), chemosensory proteins (CSPs), and β-fructofuranosidase (β-FFase).
Two-step method in OGS. First, we collected the reference protein sequences of each gene family from NCBI GenBank. And the reference protein sequences were further manually confirmed. Then, we used BLASTP to determine candidate sequences from OGS of each insect (E-value < 1E-5). Next, HMMER was used to align the candidate sequences to the Pfam database (E-value < 1E-5).
For the phylogenetic analysis of gene families
we aligned protein sequences of each gene family using MAFFT and filtered sequences with trimAl to obtain the conserved blocks.
IQ-Tree was used to construct the phylogenetic tree with the best model estimated by ModelFinder (1000 ultrafast bootstrap approximation replicates). The tree was visualized using FigTree.
An R package RIdeogram was used to map and visualize genes in chromosomes.
3.11 ZJ品系的鉴定
We identified the Tpi gene and determined the strain from the Zhejiang population using sites in the fourth exon of Tpi (TpiE4-165, TpiE4-168 and TpiE4-183).
3.12 性染色体
To identify the sex chromosomes (Z and W chromosomes) in fall armyworm, one female pupa and one male pupa were re-sequenced using Illumina HiSeq platforms to obtain an approximate 40X coverage.
The paired-end sequencing data of the female pupa was used as an input to Jellyfish with k-mer length =17 and genomescope for assessment of genomic heterozygosity and genomic size.
Normalized coverage levels of sequence reads from the Z chromosome in males should be twice that of females. In contrast, males do not have any DNA contribution from the W chromosome, while the autosomes should have equal coverage between males and females.
Thus, a difference in sequencing coverage ratio is expected for both Z and W chromosomes between sexes, but not autosomes and this difference can be used to identify sex-linked scaffolds. (利用测序深度减半的原理)
After filtering with fqtools, genome re-sequencing reads were aligned to the fall armyworm genome assembly using Bowtie2 with default parameters. Analysis and visualization of the log2 of the male:female (M:F) coverage ratio were performed using the R package ‘changepoint’ v2.2.2(https://CRAN.R-project.org/package=changepoint).
3.13 正选择分析
All 5,410 single-copy genes shared by four Noctuidae insects, S. frugiperda, S. litura, T. ni and H. armigera were used for positive selection analysis.
Protein sequences of each single-copy gene family were aligned using MAFFT v7, and then the protein alignments were converted to their corresponding nucleotide alignments by the Perl script PAL2NAL.
The dN/dS ratio was estimated for each homologous cluster using the CodeML program in the PAML package (branch-site model).
We calculated the significances of obtained positive-selected genes using the Chi-square test with a false discovery rate (FDR) cutoff of 0.05.
3.14 富集分析
The GO and KEGG enrichment analyses were conducted using Omicshare CloudTools under this tool’s default instructions (http://www.omicshare.com/).
4. 结果与讨论
4.1 草地贪夜蛾染色体组组装
There were 194 contigs (126 Mb) identified as representing allelic variants of sequence already present in the assembly and these were removed.
(1) different sequencing technologies, sequencing depth, and assembly approaches. (2) fall armyworm samples are from different areas/habitats.
It has been reported that variable genome size of different strains within the same species may be a result of the amplification, deletion and divergence of repetitive sequences; colonization of new environments; variation of environmentally-dependent life history traits.
Further study is needed to determine the reason of the genome size variation in the different strains of fall armyworm.
The assembled genome size was 155 Mb larger than the genome size estimated by 17-mer analysis.
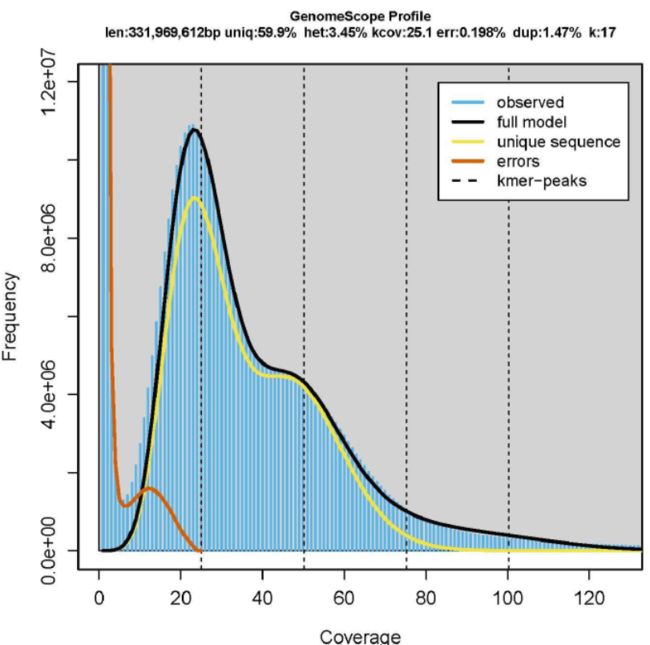
ZJ-version genome assembly had only 525 gaps and the gap lengths were estimated to be 53 kb, suggesting that the ZJ-version genome assembly was highly complete.
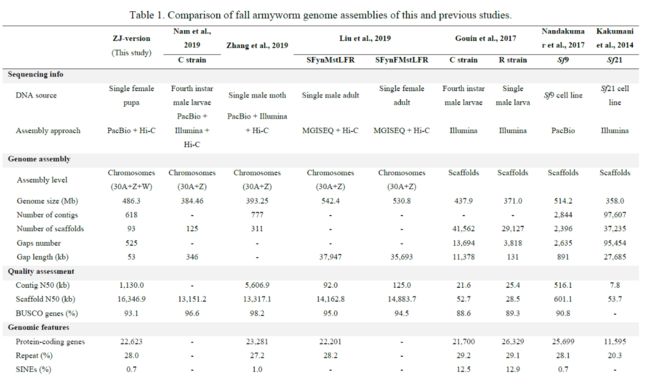
According to our Hi-C interaction information, we cut the primary assembly into 618 contigs, then anchored 556 (97.4% in length) contigs to 31 chromosomes (30 autosomes and Z chromosome).
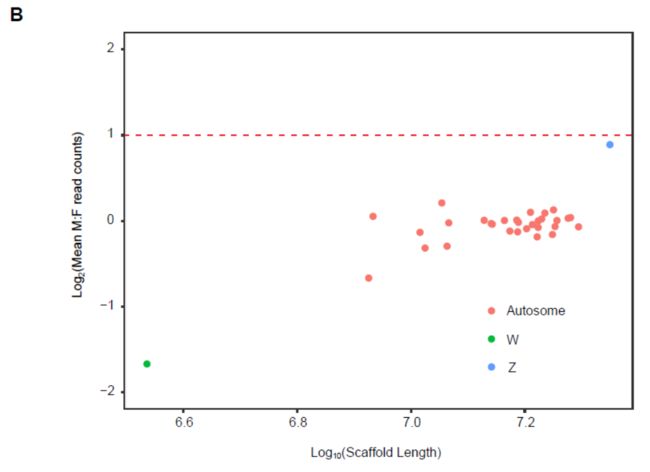
The largest super-scaffold (Chr1) yielded two-fold greater male coverage, as expected for the Z chromosome. Although we failed to obtain an intact W chromosome using Hi-C scaffolding, we have identified 4.7 Mb W-linked sequences in the unanchored contigs, including a long W-linked contig (ctg37, contig 37) of a length of 3.5 Mb.
Because the Lepidopteran W chromosome is enriched in repeat sequences, it is difficult to assemble a complete W chromosome with present sequencing and assembly methods.
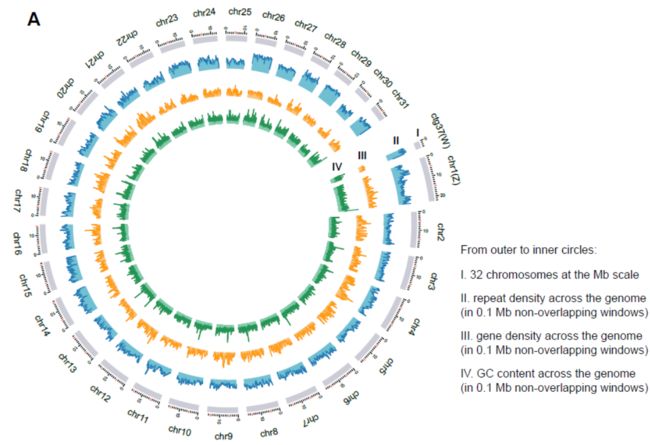
The fall armyworm genome shares high synteny with other Lepidopteran insect genomes showing a strong evidence for genome conservation at the chromosome level in Noctuidae insects.
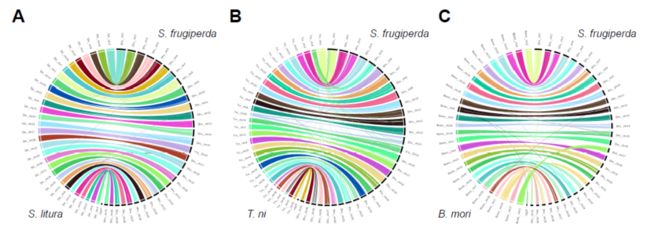
4.2 基因组注释
28% of the ZJ-version fall armyworm genome was annotated as repeat sequences.
22,623 protein-coding genes were annotated in the ZJ-version fall armyworm genome.

4.3 浙江省草地贪夜蛾为C品系
Based on these sites, we determined that the strain of fall armyworm indicated by the ZJ-version genome is the C strains, which is the same strain found in the Yunnan population.

4.4 直系同源基因与比较基因组分析
22 insect genomes covering six insect orders (Lepidoptera, Trichoptera, Diptera, Coleoptera, Hymenoptera, and Hemiptera).
gene expansion might have occurred in the fall armyworm genome.
The expansion of nutrition metabolism and transport system genes might facilitate the absorption of nutrients from different plant hosts and the detoxification of natural xenobiotics from plants.
5,410 single-copy genes were used for positive selection analyses. As a result, we identified 835 positive selected genes in the fall armyworm using the Branch-site model in PAML, including the GRs.
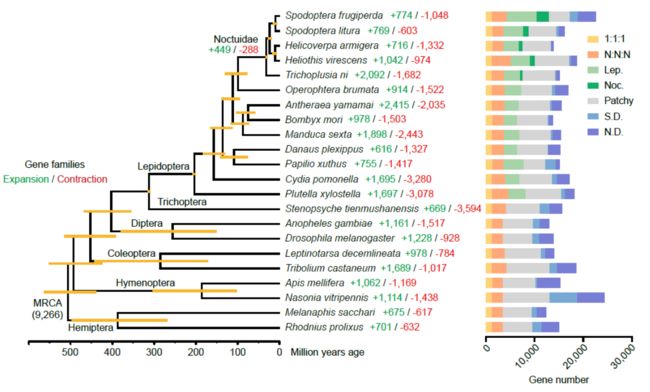
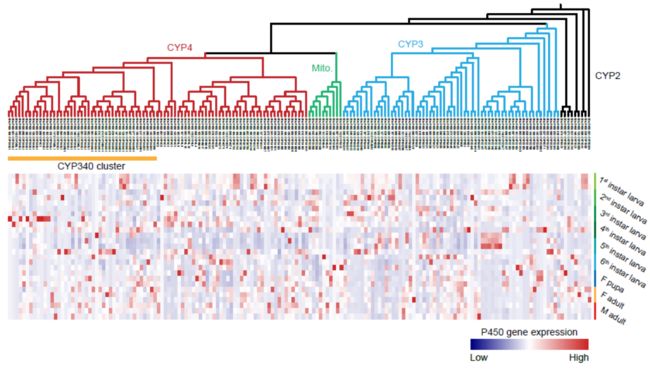
4.5 细胞色素P450基因家族基因扩张和广泛表达
P450 clans 3 and 4 show a large expansion in the fall armyworm comparing with that in the model insect of Lepidoptera, the silkworm. However, P450 clans Mito and 2 were strongly conserved between the fall armyworm and silkworm.
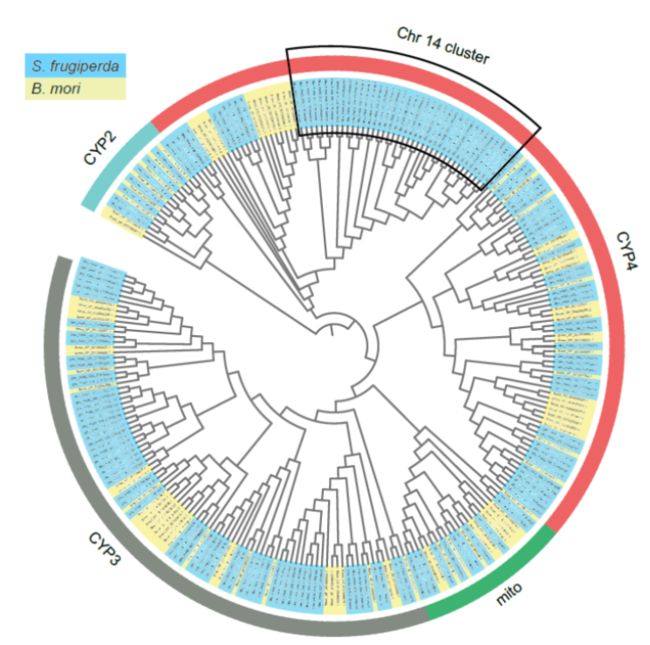
A total of 163 P450 genes were mapped to the 23 chromosomes of fall armyworm. Distribution analysis showed at least 19 P450s clusters exist in the fall armyworm genome. The largest P450 cluster was located on Chr14 and consisted of 39 CYP340 genes.
P450 genes tended to be widely expressed in all developmental stages.
The expansion of P450 in clan 3 and clan 4, and the wide-spread expression of these P450 genes in almost all developmental stages are likely important for fall armyworm to detoxify the plant xenobiotics.
4.6 味觉受体的转录组学与进化分析
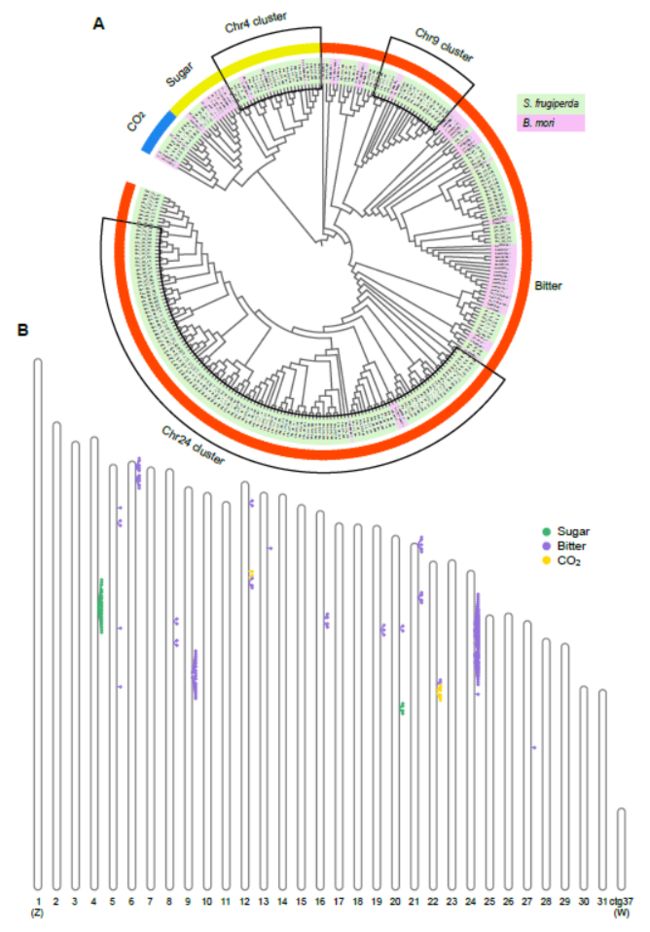
We identified 221 gustatory receptors genes which includes 189 bitter receptors, 24 sugar receptors and 8 CO2 receptors using a manual annotation pipeline.
Two large GR clusters were found on Chr9 and 24. The sugar receptors were also tandemly duplicated on chromosome 4 of the fall armyworm.
Fast host-recognition is important to maintain the energy requirements for fall armyworm in long distance migration, the expansion of GR genes likely facilitates host-recognition.
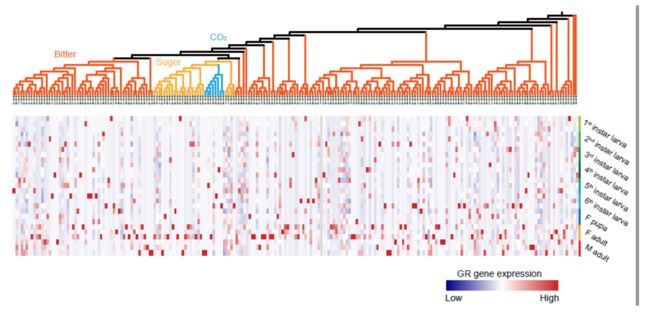
Transcriptome analysis indicated that 152 out of 189 bitter GR genes were detected as expressed genes, the GR genes tended to express in the adult.
4.7 β-呋喃果糖苷酶基因的扩增
As a sucrase, β-FFase, is responsible for cleaving sucrose to maintain cell metabolism and growth in bacteria and plants and was assumed to have not existed in animals for many years.
Recent studies show that in insects, β-FFase genes were acquired via horizontal gene transfer from bacteria and function in insect avoidance of plant secondary metabolites and glycometabolism modulation.
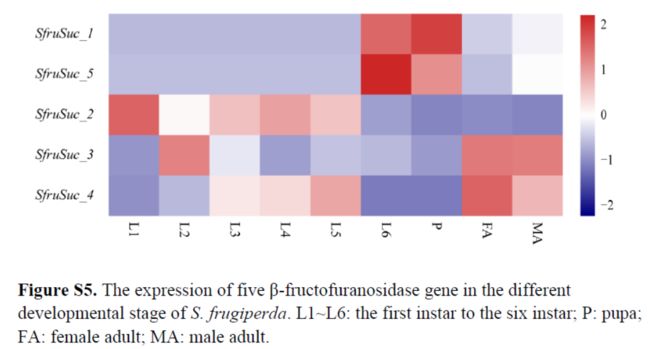
five β-FFase genes (SfruSuc1, SfruSuc2, SfruSuc3, SfruSuc4 and SfruSuc5) were identified and they exhibited significant gene expansion compared with those of the silkworm.
5. 参考文献
[1]江南纪, 王琛柱. 草地贪夜蛾的性信息素通讯研究进展[J]. 昆虫学报, 2019, 62(08): 993–1002.
[2]KaXiao, H., Ye, X., Xu, H., Mei, Y., Yang, Yi, Chen, X., Yang, Yajun, Liu, T., Yu, Y., Yang, W., Lu, Z., Li, F., 2020. The genetic adaptations of fall armyworm Spodoptera frugiperda facilitated its rapid global dispersal and invasion. Mol Ecol Resour 1755–0998.13182. https://doi.org/10/ggtqbz