最近真的是懒得要死,很多东西真的是做了没有及时整理出来,像这篇文献已经读过了,方法也去尝试过了,但是就是没有整理出来,正所谓好记性不如烂笔头。因为自己一直在分析单细胞测序的数据,总是画了聚类图后,想着怎么鉴定出细胞类型来,然后刚好读到这篇文献,讲了用CHETAH,基于已发表的文献的鉴定出来的细胞类型来鉴定,这种感觉就像是以前用已知数据来训练未知数据一样,我感觉随着单细胞测序数据越来越多,这种鉴定方法将成为一种趋势,以后应该会用更好的算法来实现,我想想自己就是个菜鸟哈哈哈,能用能看懂就很开心呢了。
一、文章总体思路
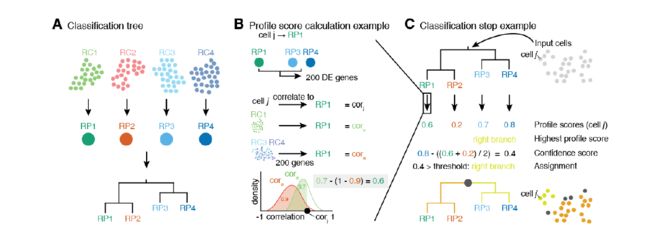
文章的思路:
A:利用已知的数据,构建一棵分类树。比如一套数据中有四种细胞类型 RP1 RP2 RP3 RP4,文章首先是计算这四种细胞类型的平均值,然后算着四种类型细胞的相关性,然后进行层次聚类,构建出分类树。
B: 拿到一个cell j,比如假定说属于RP1 类,那么久计算这个细胞和RP1的相关性,也要计算cell j与除RP1 以外的 【RP3 RP4】(将这两个当作一个整体)的相关性,为什么不计算RP2呢,因为我是这么想的,RP2 和RP1属于都一个根下,所以都加进去计算会不太好吧,具体原理不是很清楚。这里计算完之后,因为这个算法是对每个细胞选定前200个差异基因(RP1 对RP3 RP4的差异基因,文章说效果比较好)来计算相关性,因此每个细胞的200个基因是不一样,所以作者这边就计算RP1 【RP3 RP4】的理论分布,然后把相关性转化为落在理论分布的profile score,之后还计算了像p值一样的confidence score。
C:就是对每个细胞做上述过程,然后得到cell type.
细胞类型的鉴定过程取决于你所选的reference data的可靠性咯。
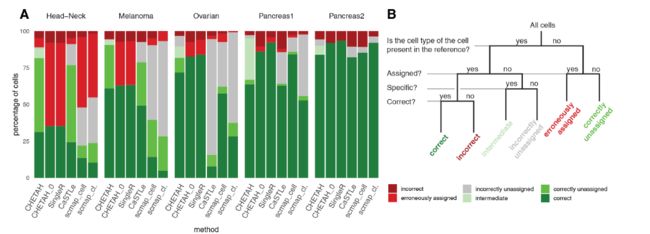
作者还比较这个算法与其他方法的结果,与之前的SingleR的结果不相上下,但是SingleR是用bulk RNA数据。
二、代码实现
1. 网址
vignette("CHETAH_introduction")

2. 输入输出
1)输入
If you have your data stored as SingleCellExperiments, continue to the next step. Otherwise, you need the following data before you begin:
- 输入你要分析的单细胞数据矩阵【 input scRNA-seq count data of the cells to be classified a data.frame or matrix, with cells in the columns and genes in the rows】
- 标准化后的参考的单细胞数据矩阵(!) 【normalized scRNA-seq count data of reference cells in similar format as the input】
- 参考的单细胞数据的细胞类型【the cell types of the reference cells
a named character vector (names corresponding to the colnames of the reference counts)】
- 可选用来可视化的要分析的单细胞数据的2维的TSNE PCA数值【(optional) a 2D reduced dimensional representation of your input data for visualization: e.g. t-SNE1, PCA,a two-column matrix/data.frame, with the cells in the rows and the two dimensions in the columns】
2)输出
CHETAH constructs a classification tree by hierarchically clustering the reference data. The classification is guided by this tree. In each node of the tree, input cells are either assigned to the right, or the left branch. A confidence score is calculated for each of these assignments. When the confidence score for an assignment is lower than the threshold (default = 0.1), the classification for that cell stops in that node.
This results in two types of classifications:
- 1.最终的细胞类型【 final types: Cells that are classified to one of the leaf nodes of the tree (i.e. a cell type of the reference)】
- 中间细胞类型,其实就没办法细分的细胞类型,也就是处于根的时候,左右两边的细胞类型的得分相似。【intermediate types: Cells that had a confidence score lower than the threshold in a certain node are assigned to that intermediate node of the tree. This happens when a cell has approximately the same similarity to the cell types in right and the left branch of that node.】
- 3.中间细胞类型在图中的显示就是Unassigned “Node1”, “Node2”等
【CHETAH generates generic names for the intermediate types: Unassigned for cells that are classified to the very first node, and “Node1”, “Node2”, etc for the additional nodes】
2.代码实例
(1)安装加载包
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("CHETAH")
vignette("CHETAH_introduction")
library(CHETAH)
(2) 构建reference data
1) 首先你需要下载你要参考的单细胞的数据,我是从Mouse Cell Atlas中下载小鼠脑的数据,然后导入R中分析
setwd("数据存的路径/Mouse Cell Atls")
adult_brain_exp <- read.csv("adult_brain_cell_exp.csv",header=T)
adult_cell_type <- read.csv("adult_brain_cell.csv",header=T)
## filter gene by a gene is expressed at least in 50 cells 因为基因数目比较多,我就把在少于50个细胞中表达的基因过滤了
row_sum <- apply(adult_brain_exp[,-1],1,sum)
filter_exp <- adult_brain_exp[row_sum>50,]#一个基因至少在50个细胞中表达
genename <- as.character(filter_exp[,1])#genename中有两个2-Mar 基因名字
final_exp <- as.matrix(filter_exp[,-1])
index <- which(genename=="2-Mar")
genename[index[1]]<- "2-Mar.1"
rownames(final_exp)<-genename
final_exp <- na.omit(final_exp) # 我数据中不知道怎么有缺失值,然后之前跑一直不成功,我试了那么久才发现
2)我把数据进行标准化和中心化,我使用seurat来做的,可以自己写代码
#Normalization and Scale data and use the Top 3000 variable gene
library(Seurat)
exp_1 <- CreateSeuratObject(counts = final_exp)
exp_1 <- SCTransform(exp_1)
exp_2 <- exp_1@[email protected]
3) 因为参考的数据里面可能不同细胞类型的数目存在差异,作者认为细胞数目在10-200之间是比较好,计算量不会太大,所以有一步选择细胞的过程。我原始数据中有一类细胞有1000多,如果不选的话,会导致构建分类树的时候,结果不准确。
# Cell selction
cell_selection <- unlist(lapply(unique(adult_cell_type$cell.type), function(type) {
type_cells <- which(adult_cell_type$cell.type == type)
if (length(type_cells) > 200) {
type_cells[sample(length(type_cells), 200)]
} else type_cells
}))
4) 构建reference data 并用自身数据进行分类看看结果
## make reference data
ref_count <- exp_2[,cell_selection]
ref_cellid <- adult_cell_type$cell.id[cell_selection]
ref_celltype <- adult_cell_type$cell.type[cell_selection]
all.equal(as.character(ref_cellid), colnames(ref_count))
adult_brain_ref <- SingleCellExperiment(assays = list(counts = ref_count),
colData = DataFrame(celltypes = ref_celltype))
#Reference QC
CorrelateReference(ref_cells = adult_brain_ref)
ClassifyReference(ref_cells = adult_brain_ref)
(3) 导入自己的数据
虽然文章没有说自己的数据需不需要标准化,但是我还是做了,我感觉没有多大影响
## input data
input_count <- readsCountSM_TSNE@assays$RNA@counts
## Normalization
input_count <- apply(input_count,2,function(column) log2((column/sum(column) * 100000) + 1))
reduced_tsne <- Embeddings(readsCountSM_TSNE, reduction = "tsne")#使用embeddings 函数来调用tsne 值
all.equal(rownames(reduced_tsne), colnames(input_count))
input_drug <- SingleCellExperiment(assays = list(counts = input_count),
reducedDims = SimpleList(TSNE = reduced_tsne))
(4)分类,然后把分类结果导入从Seurat得到的object中, 就可以了
## Classfication
input_drug <- CHETAHclassifier(input = input_drug,
ref_cells = adult_brain_ref)
PlotCHETAH(input_drug)
input_drug <- Classify(input_drug, 0)
PlotCHETAH(input = input_drug, tree = FALSE)
pdf("cell_type_analyzed_by_adult_brain_type_from_Mouse_cell_atlas.pdf",8,8)
DimPlot(object = readsCountSM_TSNE, reduction = 'tsne',label = TRUE,group.by = 'ident',shape.by="orig.ident",repel=TRUE,label.size=5,pt.size=1.5)
dev.off()
levels(readsCountSM_TSNE$orig.ident)[1]="3-HB"
levels(readsCountSM_TSNE$orig.ident)[2]="Control"
Sample_cluster(readsCountSM_TSNE)
3. 总结
我感觉这个方法还是不错的,结果还是挺准的。但是还是取决于所参考的数据。