- 机器学习与深度学习间关系与区别
ℒℴѵℯ心·动ꦿ໊ོ꫞
人工智能学习深度学习python
一、机器学习概述定义机器学习(MachineLearning,ML)是一种通过数据驱动的方法,利用统计学和计算算法来训练模型,使计算机能够从数据中学习并自动进行预测或决策。机器学习通过分析大量数据样本,识别其中的模式和规律,从而对新的数据进行判断。其核心在于通过训练过程,让模型不断优化和提升其预测准确性。主要类型1.监督学习(SupervisedLearning)监督学习是指在训练数据集中包含输入
- 铭刻于星(四十二)
随风至
69夜晚,绍敏同学做完功课后,看了眼房外,没听到动静才敢从书包的夹层里拿出那个心形纸团。折痕压得很深,都有些旧了,想来是已经写好很久了。绍敏同学慢慢地、轻轻地捏开折叠处,待到全部拆开后,又反复抚平纸张,然后仔细地一字字默看。只是开头的三个字是第一次看到,让她心漏跳了几拍。“亲爱的绍敏:从四年级的时候,我就喜欢你了,但是我一直不敢说,怕影响你学习。六年级的时候听说有人跟你表白,你接受了,我很难过,但
- 《策划经理回忆录之二》
路基雅虎
话说三年变六年,飘了,飘了……眨眼,2013年5月,老吴回到了他的家乡——油城从新开启他的工作幻想症生涯。很庆幸,这是一家很有追求,同时敢于尝试的,且实力不容低调的新星房企——金源置业(前身泰源置业)更值得庆幸的是第一个盘就是油城十路的标杆之一:金源盛世。2013年5月,到2015年11月,两年的陪伴,迎来了一场大爆发。2000个筹,5万/筹,直接回笼1个亿!!!这……让我开始认真审视这座看似五线
- 放下是一段成长的修行
小莳玥
人来到这个世界上,只有两件事:生和死。一件事已经做完了,另一件你还急什么呢?是人,都有七情六欲。是心,都有喜怒哀乐,这些再正常不过了。别总抱怨自己活得累,过得辛苦。永远记住:舒坦是留给死人的。苦,才是生活;累,才是工作;变,才是命运;忍,才是历练;容,才是智慧;静,才是修养;舍,才会得到;做,才会拥有。人生,活得太清楚,才是最大的不明白。有些事,看得很清,却说不清;有些人,了解很深,却猜不透;有些
- 第四天旅游线路预览——从换乘中心到喀纳斯湖
陟彼高冈yu
基于Googleearthstudio的旅游规划和预览旅游
第四天:从贾登峪到喀纳斯风景区入口,晚上住宿贾登峪;换乘中心有4路车,喀纳斯①号车,去喀纳斯湖,路程时长约5分钟;将上面的的行程安排进行动态展示,具体步骤见”Googleearthstudio进行动态轨迹显示制作过程“、“Googleearthstudio入门教程”和“Googleearthstudio进阶教程“相关内容,得到行程如下所示:Day4-2-480p
- linux中sdl的使用教程,sdl使用入门
Melissa Corvinus
linux中sdl的使用教程
本文通过一个简单示例讲解SDL的基本使用流程。示例中展示一个窗口,窗口里面有个随机颜色快随机移动。当我们鼠标点击关闭按钮时间窗口关闭。基本步骤如下:1.初始化SDL并创建一个窗口。SDL_Init()初始化SDL_CreateWindow()创建窗口2.纹理渲染存储RGB和存储纹理的区别:比如一个从左到右由红色渐变到蓝色的矩形,用存储RGB的话就需要把矩形中每个点的具体颜色值存储下来;而纹理只是一
- PHP环境搭建详细教程
好看资源平台
前端php
PHP是一个流行的服务器端脚本语言,广泛用于Web开发。为了使PHP能够在本地或服务器上运行,我们需要搭建一个合适的PHP环境。本教程将结合最新资料,介绍在不同操作系统上搭建PHP开发环境的多种方法,包括Windows、macOS和Linux系统的安装步骤,以及本地和Docker环境的配置。1.PHP环境搭建概述PHP环境的搭建主要分为以下几类:集成开发环境:例如XAMPP、WAMP、MAMP,这
- Day1笔记-Python简介&标识符和关键字&输入输出
~在杰难逃~
Pythonpython开发语言大数据数据分析数据挖掘
大家好,从今天开始呢,杰哥开展一个新的专栏,当然,数据分析部分也会不定时更新的,这个新的专栏主要是讲解一些Python的基础语法和知识,帮助0基础的小伙伴入门和学习Python,感兴趣的小伙伴可以开始认真学习啦!一、Python简介【了解】1.计算机工作原理编程语言就是用来定义计算机程序的形式语言。我们通过编程语言来编写程序代码,再通过语言处理程序执行向计算机发送指令,让计算机完成对应的工作,编程
- Python快速入门 —— 第三节:类与对象
孤华暗香
Python快速入门python开发语言
第三节:类与对象目标:了解面向对象编程的基础概念,并学会如何定义类和创建对象。内容:类与对象:定义类:class关键字。类的构造函数:__init__()。类的属性和方法。对象的创建与使用。示例:classStudent:def__init__(self,name,age,major):self.name&#
- 2023-10-22
奥雷里亚诺第n
昨天在B站看到关于猫喜欢挠人的视频,视频教导说猫挠人的话就抓住它的后脖颈然后用手打打挠人的那个爪子。视频本身没什么,但评论区却炸开了锅(真是符合挑食者厌食心理)。令我印象最深刻的一个甚至上升到了关于我是谁这种终极问题。它说,猫就是畜生,它挠人就打它别惯着它,反正我六道轮回成了人就应该保持人的高贵,谁都别想来打破。我顿时汗颜,但看到下面全是类似的言论只不过后面的理由各有不同,本来想骂人的心都凉了一半
- 数据仓库——维度表一致性
墨染丶eye
背诵数据仓库
数据仓库基础笔记思维导图已经整理完毕,完整连接为:数据仓库基础知识笔记思维导图维度一致性问题从逻辑层面来看,当一系列星型模型共享一组公共维度时,所涉及的维度称为一致性维度。当维度表存在不一致时,短期的成功难以弥补长期的错误。维度时确保不同过程中信息集成起来实现横向钻取货活动的关键。造成横向钻取失败的原因维度结构的差别,因为维度的差别,分析工作涉及的领域从简单到复杂,但是都是通过复杂的报表来弥补设计
- 第六集如何安装CentOS7.0,3分钟学会centos7安装教程
date分享
从光盘引导系统按回车键继续进入引导程序安装界面,选择语言这里选择简体中文版点击继续选择桌面安装下面给系统分区选择磁盘,点击完成选择基本分区,点击加号swap分区,大小填内存的两倍在选择根分区,使用所有可用的磁盘空间选择文件系统ext4点击完成,点击开始安装设置root密码,点击完成设置普通用户和密码,点击完成整个过程持续八分钟左右根据个人配置不同,时间长短不同好,现在点击重启系统进入重启状态点击本
- 2018-12-29
枫叶红时总多离别
2018年12月29日星期六昨天老师就告诉我们,今天下午不用上课,是图书漂流活动会。我觉得很兴奋,好期待。到了下午,我帮好忙就到外面去买书,刚一出去,就有一大帮的大哥哥、大姐姐围着我问要不要买书,买一本书送一颗糖。我看到了一本《小老虎比上树》的书,问大姐姐多少钱,大姐姐说这本书原价13块,现在便宜4块钱也就是9块钱卖给你,我就把一张10块钱给她找,她找了我一块钱。我现在想想我今天只带了10块钱,现
- C++菜鸟教程 - 从入门到精通 第二节
DreamByte
c++
一.上节课的补充(数据类型)1.前言继上节课,我们主要讲解了输入,输出和运算符,我们现在来补充一下数据类型的知识上节课遗漏了这个知识点,非常的抱歉顺便说一下,博主要上高中了,更新会慢,2-4周更新一次对了,正好赶上中秋节,小编跟大家说一句:中秋节快乐!2.int类型上节课,我们其实只用了int类型int类型,是整数类型,它们存贮的是整数,不能存小数(浮点数)定义变量的方式很简单inta;//定义一
- mac电脑命令行获取电量
小米人er
我的博客macos命令行
在macOS上,有几个命令行工具可以用来获取电量信息,最常用的是pmset命令。你可以通过以下方式来查看电池状态和电量信息:查看电池状态:pmset-gbatt这个命令会返回类似下面的输出:Nowdrawingfrom'BatteryPower'-InternalBattery-0(id=1234567)95%;discharging;4:02remainingpresent:true输出中包括电
- 简介Shell、zsh、bash
zhaosuningsn
Shellzshbashshelllinuxbash
Shell是Linux和Unix的外壳,类似衣服,负责外界与Linux和Unix内核的交互联系。例如接收终端用户及各种应用程序的命令,把接收的命令翻译成内核能理解的语言,传递给内核,并把内核处理接收的命令的结果返回给外界,即Shell是外界和内核沟通的桥梁或大门。Linux和Unix提供了多种Shell,其中有种bash,当然还有其他好多种。Mac电脑中不但有bash,还有一个zsh,预装的,据说
- 如何选择最适合你的项目研发管理软件?TAPD卓越版全面解析
北京云巴巴信息技术有限公司
产品经理需求分析
在当今快速发展的科技时代,项目研发管理软件已成为企业不可或缺的重要工具。面对市场上琳琅满目的产品,如何选择一款适合自己团队的项目研发管理软件呢?本文将围绕项目研发管理软件的选择标准,重点介绍TAPD卓越版的特点、优势以及使用体验,让你更好地理解和选择适合自己的项目研发管理软件。项目研发管理软件的选择标准在选择项目研发管理软件时,我们需要考虑以下几个方面的因素:功能全面性:软件是否覆盖了从需求管理、
- 舜公郑金锋书辛丑自剪扇面书法作品(四O六)
舜公郑金锋
辛丑小阳春,新自剪扇面400品,大多为各色撒金、撒银、描金、描银、水印、彩绘、荧光等亚粉、色宣纸,以及域外包装填充纸等;王一品长锋羊毫秃笔;一得阁云头艳墨、宿墨、水等。书体有甲骨文,金文(商周金文、春秋战国金文、中山王厝器金文、汉金文……),楚简帛书,侯马盟书,温县盟书,小篆,果蝙书等,隶书(秦简、汉简帛书、汉碑……),草书(章草、小草、大草……),行书(行楷、行草),楷书(魏碑及北朝墓志、隋朝墓
- 2019-01-19
王小康KK
姓名:王康公司:扬州市方圆建筑工程有限公司2018年3月16日~3月18日上海361期《六项精进》感谢二组学员【日精进打卡第307天】【知~学习】《六项精进》大纲3遍共862遍《大学》通篇3遍共860遍《六项精进》全书40页【经典名句】思想决定行为,行为决定习惯,习惯决定性格,性格决定命运。【行~实践】一、修身:(对自己个人)1、践行六项精进的理念。二、齐家:(对家庭和家人)1、和女朋友视频聊天。
- OPENAIGC开发者大赛企业组AI黑马奖 | AIGC数智传媒解决方案
RPA中国
人工智能AIGC传媒
在第二届拯救者杯OPENAIGC开发者大赛中,涌现出一批技术突出、创意卓越的作品。为了让这些优秀项目被更多人看到,我们特意开设了优秀作品报道专栏,旨在展示其独特之处和开发者的精彩故事。无论您是技术专家还是爱好者,希望能带给您不一样的知识和启发。让我们一起探索AIGC的无限可能,见证科技与创意的完美融合!创未来AI应用赛-企业组AI黑马奖作品名称:AIGC数智传媒解决方案参赛团队:深圳市三象智能技术
- 【六】阿伟开始搭建Kafka学习环境
能源恒观
中间件学习kafkaspring
阿伟开始搭建Kafka学习环境概述上一篇文章阿伟学习了Kafka的核心概念,并且把市面上流行的消息中间件特性进行了梳理和对比,方便大家在学习过程中进行对比学习,最后梳理了一些Kafka使用中经常遇到的Kafka难题以及解决思路,经过上一篇的学习我相信大家对Kafka有了初步的认识,本篇将继续学习Kafka。一、安装和配置学习一项技术首先要搭建一套服务,而Kafka的运行主要需要部署jdk、zook
- 2018-12-07
旅一06丁琪琪
旅管一班6组学号链接06丁琪琪16家国鑫26孟令慧30王思宁36温红丽46朱赵筱楠
- STM32中的计时与延时
lupinjia
STM32stm32单片机
前言在裸机开发中,延时作为一种规定循环周期的方式经常被使用,其中尤以HAL库官方提供的HAL_Delay为甚。刚入门的小白可能会觉得既然有官方提供的延时函数,而且精度也还挺好,为什么不用呢?实际上HAL_Delay中有不少坑,而这些也只是HAL库中无数坑的其中一些。想从坑里跳出来还是得加强外设原理的学习和理解,切不可只依赖HAL库。除了延时之外,我们在开发中有时也会想要确定某段程序的耗时,这就需要
- 安徽省这个湖,比西湖大8倍,称是安徽的北戴河, 合肥的后花园
旅游小号角
旅游爱好者都知道,安徽省是一个旅游资源十分丰富的省份,且不说黄山、九华山、天柱山这三大名山,单说湖泊就不比其它省份少,今天我们一起走遍世界将为大家说说一个号称安徽北戴河,合肥后花园的湖泊,看看到底是哪个湖泊?话说,这个湖泊位于安徽省六安市舒城县境内,东距合肥50千米,大约一个小时左右的车程,它号称是合肥的后花园,安徽的北戴河。相传,湖畔石壁之上有一奇石神似观音临湖,湖中漂动众多小岛栩栩如佛子,宛若
- 简单了解 JVM
记得开心一点啊
jvm
目录♫什么是JVM♫JVM的运行流程♫JVM运行时数据区♪虚拟机栈♪本地方法栈♪堆♪程序计数器♪方法区/元数据区♫类加载的过程♫双亲委派模型♫垃圾回收机制♫什么是JVMJVM是JavaVirtualMachine的简称,意为Java虚拟机。虚拟机是指通过软件模拟的具有完整硬件功能的、运行在一个完全隔离的环境中的完整计算机系统(如:JVM、VMwave、VirtualBox)。JVM和其他两个虚拟机
- 最超值的Mac——Mac mini
初心么么哒
你知道最超值的Mac是什么吗?自2005年以来,Macmini一直是Apple台式机产品线中的主要产品。最初推出是为了让对Mac好奇的Mac进入Apple生态系统的一种简单方式,现在新的AppleSiliconMacmini可能是任何寻找新Mac的人的最有吸引力的购买。什么是AppleSiliconMacmini?M1Macmini是Apple最小的台式电脑,同时也是最快的台式电脑之一。最新型号由
- 一文掌握python面向对象魔术方法(二)
程序员neil
pythonpython开发语言
接上篇:一文掌握python面向对象魔术方法(一)-CSDN博客目录六、迭代和序列化:1、__iter__(self):定义迭代器,使得类可以被for循环迭代。2、__getitem__(self,key):定义索引操作,如obj[key]。3、__setitem__(self,key,value):定义赋值操作,如obj[key]=value。4、__delitem__(self,key):定义
- Python实现简单的机器学习算法
master_chenchengg
pythonpython办公效率python开发IT
Python实现简单的机器学习算法开篇:初探机器学习的奇妙之旅搭建环境:一切从安装开始必备工具箱第一步:安装Anaconda和JupyterNotebook小贴士:如何配置Python环境变量算法初体验:从零开始的Python机器学习线性回归:让数据说话数据准备:从哪里找数据编码实战:Python实现线性回归模型评估:如何判断模型好坏逻辑回归:从分类开始理论入门:什么是逻辑回归代码实现:使用skl
- 《度五行》生活报报甲午62:不通痛苦,太通也痛苦,要健康快乐,需要通体舒畅。
YangduSam2021
220809壬寅戊申甲午,《度.生活五行》:天干土克水,水生木,木克土。地支寅申冲,寅午合。20220809,周二,兴大上海六班2512天,西交大2013上海班3212天,后TA15332天,度生活619天,今天拜访了一家有趣且当红产业的新创公司AK。AK一开始从事深海新能源储存与供电设备的研发生产制造,2年前开始做移动与家庭储能设备的研发生产制造。觉得有趣是因为这是笔者认知里用科技做降维打击的公
- JVM、JRE和 JDK:理解Java开发的三大核心组件
Y雨何时停T
Javajava
Java是一门跨平台的编程语言,它的成功离不开背后强大的运行环境与开发工具的支持。在Java的生态中,JVM(Java虚拟机)、JRE(Java运行时环境)和JDK(Java开发工具包)是三个至关重要的核心组件。本文将探讨JVM、JDK和JRE的区别,帮助你更好地理解Java的运行机制。1.JVM:Java虚拟机(JavaVirtualMachine)什么是JVM?JVM,即Java虚拟机,是Ja
- 关于旗正规则引擎下载页面需要弹窗保存到本地目录的问题
何必如此
jsp超链接文件下载窗口
生成下载页面是需要选择“录入提交页面”,生成之后默认的下载页面<a>标签超链接为:<a href="<%=root_stimage%>stimage/image.jsp?filename=<%=strfile234%>&attachname=<%=java.net.URLEncoder.encode(file234filesourc
- 【Spark九十八】Standalone Cluster Mode下的资源调度源代码分析
bit1129
cluster
在分析源代码之前,首先对Standalone Cluster Mode的资源调度有一个基本的认识:
首先,运行一个Application需要Driver进程和一组Executor进程。在Standalone Cluster Mode下,Driver和Executor都是在Master的监护下给Worker发消息创建(Driver进程和Executor进程都需要分配内存和CPU,这就需要Maste
- linux上独立安装部署spark
daizj
linux安装spark1.4部署
下面讲一下linux上安装spark,以 Standalone Mode 安装
1)首先安装JDK
下载JDK:jdk-7u79-linux-x64.tar.gz ,版本是1.7以上都行,解压 tar -zxvf jdk-7u79-linux-x64.tar.gz
然后配置 ~/.bashrc&nb
- Java 字节码之解析一
周凡杨
java字节码javap
一: Java 字节代码的组织形式
类文件 {
OxCAFEBABE ,小版本号,大版本号,常量池大小,常量池数组,访问控制标记,当前类信息,父类信息,实现的接口个数,实现的接口信息数组,域个数,域信息数组,方法个数,方法信息数组,属性个数,属性信息数组
}
&nbs
- java各种小工具代码
g21121
java
1.数组转换成List
import java.util.Arrays;
Arrays.asList(Object[] obj); 2.判断一个String型是否有值
import org.springframework.util.StringUtils;
if (StringUtils.hasText(str)) 3.判断一个List是否有值
import org.spring
- 加快FineReport报表设计的几个心得体会
老A不折腾
finereport
一、从远程服务器大批量取数进行表样设计时,最好按“列顺序”取一个“空的SQL语句”,这样可提高设计速度。否则每次设计时模板均要从远程读取数据,速度相当慢!!
二、找一个富文本编辑软件(如NOTEPAD+)编辑SQL语句,这样会很好地检查语法。有时候带参数较多检查语法复杂时,结合FineReport中生成的日志,再找一个第三方数据库访问软件(如PL/SQL)进行数据检索,可以很快定位语法错误。
- mysql linux启动与停止
墙头上一根草
如何启动/停止/重启MySQL一、启动方式1、使用 service 启动:service mysqld start2、使用 mysqld 脚本启动:/etc/inint.d/mysqld start3、使用 safe_mysqld 启动:safe_mysqld&二、停止1、使用 service 启动:service mysqld stop2、使用 mysqld 脚本启动:/etc/inin
- Spring中事务管理浅谈
aijuans
spring事务管理
Spring中事务管理浅谈
By Tony Jiang@2012-1-20 Spring中对事务的声明式管理
拿一个XML举例
[html]
view plain
copy
print
?
<?xml version="1.0" encoding="UTF-8"?>&nb
- php中隐形字符65279(utf-8的BOM头)问题
alxw4616
php中隐形字符65279(utf-8的BOM头)问题
今天遇到一个问题. php输出JSON 前端在解析时发生问题:parsererror.
调试:
1.仔细对比字符串发现字符串拼写正确.怀疑是 非打印字符的问题.
2.逐一将字符串还原为unicode编码. 发现在字符串头的位置出现了一个 65279的非打印字符.
- 调用对象是否需要传递对象(初学者一定要注意这个问题)
百合不是茶
对象的传递与调用技巧
类和对象的简单的复习,在做项目的过程中有时候不知道怎样来调用类创建的对象,简单的几个类可以看清楚,一般在项目中创建十几个类往往就不知道怎么来看
为了以后能够看清楚,现在来回顾一下类和对象的创建,对象的调用和传递(前面写过一篇)
类和对象的基础概念:
JAVA中万事万物都是类 类有字段(属性),方法,嵌套类和嵌套接
- JDK1.5 AtomicLong实例
bijian1013
javathreadjava多线程AtomicLong
JDK1.5 AtomicLong实例
类 AtomicLong
可以用原子方式更新的 long 值。有关原子变量属性的描述,请参阅 java.util.concurrent.atomic 包规范。AtomicLong 可用在应用程序中(如以原子方式增加的序列号),并且不能用于替换 Long。但是,此类确实扩展了 Number,允许那些处理基于数字类的工具和实用工具进行统一访问。
- 自定义的RPC的Java实现
bijian1013
javarpc
网上看到纯java实现的RPC,很不错。
RPC的全名Remote Process Call,即远程过程调用。使用RPC,可以像使用本地的程序一样使用远程服务器上的程序。下面是一个简单的RPC 调用实例,从中可以看到RPC如何
- 【RPC框架Hessian一】Hessian RPC Hello World
bit1129
Hello world
什么是Hessian
The Hessian binary web service protocol makes web services usable without requiring a large framework, and without learning yet another alphabet soup of protocols. Because it is a binary p
- 【Spark九十五】Spark Shell操作Spark SQL
bit1129
shell
在Spark Shell上,通过创建HiveContext可以直接进行Hive操作
1. 操作Hive中已存在的表
[hadoop@hadoop bin]$ ./spark-shell
Spark assembly has been built with Hive, including Datanucleus jars on classpath
Welcom
- F5 往header加入客户端的ip
ronin47
when HTTP_RESPONSE {if {[HTTP::is_redirect]}{ HTTP::header replace Location [string map {:port/ /} [HTTP::header value Location]]HTTP::header replace Lo
- java-61-在数组中,数字减去它右边(注意是右边)的数字得到一个数对之差. 求所有数对之差的最大值。例如在数组{2, 4, 1, 16, 7, 5,
bylijinnan
java
思路来自:
http://zhedahht.blog.163.com/blog/static/2541117420116135376632/
写了个java版的
public class GreatestLeftRightDiff {
/**
* Q61.在数组中,数字减去它右边(注意是右边)的数字得到一个数对之差。
* 求所有数对之差的最大值。例如在数组
- mongoDB 索引
开窍的石头
mongoDB索引
在这一节中我们讲讲在mongo中如何创建索引
得到当前查询的索引信息
db.user.find(_id:12).explain();
cursor: basicCoursor 指的是没有索引
&
- [硬件和系统]迎峰度夏
comsci
系统
从这几天的气温来看,今年夏天的高温天气可能会维持在一个比较长的时间内
所以,从现在开始准备渡过炎热的夏天。。。。
每间房屋要有一个落地电风扇,一个空调(空调的功率和房间的面积有密切的关系)
坐的,躺的地方要有凉垫,床上要有凉席
电脑的机箱
- 基于ThinkPHP开发的公司官网
cuiyadll
行业系统
后端基于ThinkPHP,前端基于jQuery和BootstrapCo.MZ 企业系统
轻量级企业网站管理系统
运行环境:PHP5.3+, MySQL5.0
系统预览
系统下载:http://www.tecmz.com
预览地址:http://co.tecmz.com
各种设备自适应
响应式的网站设计能够对用户产生友好度,并且对于
- Transaction and redelivery in JMS (JMS的事务和失败消息重发机制)
darrenzhu
jms事务承认MQacknowledge
JMS Message Delivery Reliability and Acknowledgement Patterns
http://wso2.com/library/articles/2013/01/jms-message-delivery-reliability-acknowledgement-patterns/
Transaction and redelivery in
- Centos添加硬盘完全教程
dcj3sjt126com
linuxcentoshardware
Linux的硬盘识别:
sda 表示第1块SCSI硬盘
hda 表示第1块IDE硬盘
scd0 表示第1个USB光驱
一般使用“fdisk -l”命
- yii2 restful web服务路由
dcj3sjt126com
PHPyii2
路由
随着资源和控制器类准备,您可以使用URL如 http://localhost/index.php?r=user/create访问资源,类似于你可以用正常的Web应用程序做法。
在实践中,你通常要用美观的URL并采取有优势的HTTP动词。 例如,请求POST /users意味着访问user/create动作。 这可以很容易地通过配置urlManager应用程序组件来完成 如下所示
- MongoDB查询(4)——游标和分页[八]
eksliang
mongodbMongoDB游标MongoDB深分页
转载请出自出处:http://eksliang.iteye.com/blog/2177567 一、游标
数据库使用游标返回find的执行结果。客户端对游标的实现通常能够对最终结果进行有效控制,从shell中定义一个游标非常简单,就是将查询结果分配给一个变量(用var声明的变量就是局部变量),便创建了一个游标,如下所示:
> var
- Activity的四种启动模式和onNewIntent()
gundumw100
android
Android中Activity启动模式详解
在Android中每个界面都是一个Activity,切换界面操作其实是多个不同Activity之间的实例化操作。在Android中Activity的启动模式决定了Activity的启动运行方式。
Android总Activity的启动模式分为四种:
Activity启动模式设置:
<acti
- 攻城狮送女友的CSS3生日蛋糕
ini
htmlWebhtml5csscss3
在线预览:http://keleyi.com/keleyi/phtml/html5/29.htm
代码如下:
<!DOCTYPE html>
<html>
<head>
<meta charset="UTF-8">
<title>攻城狮送女友的CSS3生日蛋糕-柯乐义<
- 读源码学Servlet(1)GenericServlet 源码分析
jzinfo
tomcatWebservlet网络应用网络协议
Servlet API的核心就是javax.servlet.Servlet接口,所有的Servlet 类(抽象的或者自己写的)都必须实现这个接口。在Servlet接口中定义了5个方法,其中有3个方法是由Servlet 容器在Servlet的生命周期的不同阶段来调用的特定方法。
先看javax.servlet.servlet接口源码:
package
- JAVA进阶:VO(DTO)与PO(DAO)之间的转换
snoopy7713
javaVOHibernatepo
PO即 Persistence Object VO即 Value Object
VO和PO的主要区别在于: VO是独立的Java Object。 PO是由Hibernate纳入其实体容器(Entity Map)的对象,它代表了与数据库中某条记录对应的Hibernate实体,PO的变化在事务提交时将反应到实际数据库中。
实际上,这个VO被用作Data Transfer
- mongodb group by date 聚合查询日期 统计每天数据(信息量)
qiaolevip
每天进步一点点学习永无止境mongodb纵观千象
/* 1 */
{
"_id" : ObjectId("557ac1e2153c43c320393d9d"),
"msgType" : "text",
"sendTime" : ISODate("2015-06-12T11:26:26.000Z")
- java之18天 常用的类(一)
Luob.
MathDateSystemRuntimeRundom
System类
import java.util.Properties;
/**
* System:
* out:标准输出,默认是控制台
* in:标准输入,默认是键盘
*
* 描述系统的一些信息
* 获取系统的属性信息:Properties getProperties();
*
*
*
*/
public class Sy
- maven
wuai
maven
1、安装maven:解压缩、添加M2_HOME、添加环境变量path
2、创建maven_home文件夹,创建项目mvn_ch01,在其下面建立src、pom.xml,在src下面简历main、test、main下面建立java文件夹
3、编写类,在java文件夹下面依照类的包逐层创建文件夹,将此类放入最后一级文件夹
4、进入mvn_ch01
4.1、mvn compile ,执行后会在

 image.png
image.png image.png
image.png image.png
image.png image.png
image.png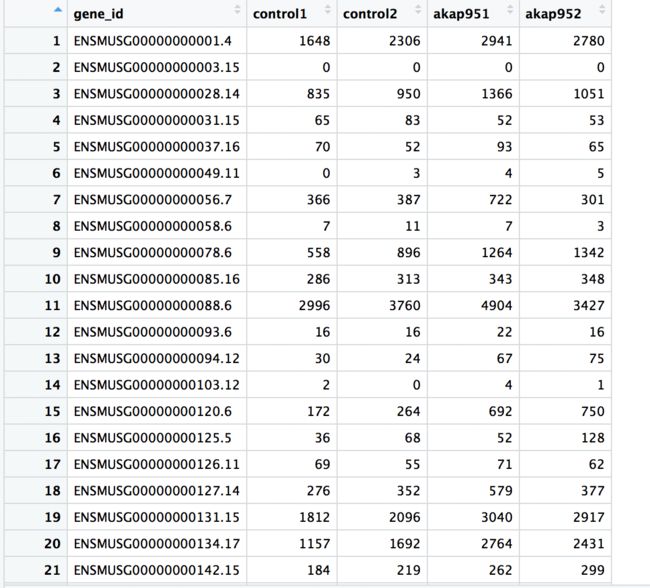 image.png
image.png image.png
image.png image.png
image.png