介绍
GDCRNATools 是一个R包,提供标准的,易于使用和全面的管道,用于下载,组织和综合分析GDC门户中的RNA表达数据,重点是解读癌症中lncRNA-mRNA相关的ceRNA调控网络。
许多分析可以使用 GDCRNATools,包括差异基因表达分析(limma(Ritchie等人2015), edgeR(Robinson,McCarthy和Smyth 2010)和 DESeq2(Love,Huber和Anders 2014)),单变量生存分析( CoxPH和KM),竞争内源RNA网络分析(超几何测试,Pearson相关分析,调节相似性分析,灵敏度Pearson偏相关)和功能富集分析(GO,KEGG,DO)。除了一些常规的可视化方法,如火山图,散点图和气泡图等,GDCRNATools中开发了三个简单的闪亮应用程序,允许用户在本地网页上显示结果。
这个用户友好的软件包允许研究人员通过简单运行一些功能并集成他们自己的管道进行分析,如分子亚型分类, 加权相关网络分析(WGCNA)(Langfelder和Horvath 2008),以及TF-miRNA共同调控网络分析等,轻松进入工作流程。
简介
GDCRNATools
GDCRNATools is an R package which provides a standard,
easy-to-use and comprehensive pipeline for downloading,
organizing, and integrative analyzing RNA expression data in the GDC portal with an emphasis on deciphering the lncRNA-mRNA related ceRNAs regulatory network in cancer. Here we provide code of the basic steps for data analysis by GDCRNATools. Detailed instructions can be found here:
http://htmlpreview.github.io/?https://github.com/Jialab-UCR/Jialab-UCR.github.io/blob/master/GDCRNATools_manual.html
1. GDCRNATools package installation
# Get the current working directory, make sure that it is
# writable, otherwise, change to a new directory
getwd()
#setwd(workingDirectory)
# installation of GDCRNATools from Bioconductor
source("https://bioconductor.org/biocLite.R")
biocLite("GDCRNATools")
library(GDCRNATools)
2. Quick start
A small internal dataset is used here to show the most basic steps for ceRNAs network analysis in GDCRNATools
2.1 Normalization of HTSeq-Counts data
### load RNA counts data
data(rnaCounts)
rnaCounts[1:5,1:5]
### load miRNAs counts data
data(mirCounts)
mirCounts[1:5,1:5]
### Normalization of RNAseq data
rnaExpr <- gdcVoomNormalization(counts = rnaCounts, filter = FALSE)
rnaExpr[1:5,1:5]
### Normalization of miRNAs data
mirExpr <- gdcVoomNormalization(counts = mirCounts, filter = FALSE)
mirExpr[1:5,1:5]
2.2 Parse and filter RNAseq metadata
metaMatrix.RNA <- gdcParseMetadata(project.id = 'TCGA-CHOL',
data.type = 'RNAseq',
write.meta = FALSE)
metaMatrix.RNA <- gdcFilterDuplicate(metaMatrix.RNA)
metaMatrix.RNA <- gdcFilterSampleType(metaMatrix.RNA)
metaMatrix.RNA[1:5,]
2.3 ceRNAs network analysis
### Identification of differentially expressed genes ###
DEGAll <- gdcDEAnalysis(counts = rnaCounts,
group = metaMatrix.RNA$sample_type,
comparison = 'PrimaryTumor-SolidTissueNormal',
method = 'limma')
DEGAll[1:5,]
### All DEGs
deALL <- gdcDEReport(deg = DEGAll, gene.type = 'all')
deALL[1:5,]
### DE long-noncoding genes
deLNC <- gdcDEReport(deg = DEGAll, gene.type = 'long_non_coding')
deLNC[1:5,]
### DE protein coding genes
dePC <- gdcDEReport(deg = DEGAll, gene.type = 'protein_coding')
dePC[1:5,]
### ceRNAs network analysis of DEGs
ceOutput <- gdcCEAnalysis(lnc = rownames(deLNC),
pc = rownames(dePC),
lnc.targets = 'starBase',
pc.targets = 'starBase',
rna.expr = rnaExpr,
mir.expr = mirExpr)
ceOutput[1:5,]
### Export ceRNAs network to Cytoscape
ceOutput2 <- ceOutput[ceOutput$hyperPValue<0.01
& ceOutput$corPValue<0.01 & ceOutput$regSim != 0,]
###### Export edges
edges <- gdcExportNetwork(ceNetwork = ceOutput2, net = 'edges')
edges[1:5,]
##### Export nodes
nodes <- gdcExportNetwork(ceNetwork = ceOutput2, net = 'nodes')
nodes[1:5,]
3. Case study: TCGA-CHOL
3.1 Download data
# set up directories for downloaded data
project <- 'TCGA-CHOL'
rnadir <- paste(project, 'RNAseq', sep='/')
mirdir <- paste(project, 'miRNAs', sep='/')
### Download RNAseq data
gdcRNADownload(project.id = 'TCGA-CHOL',
data.type = 'RNAseq',
write.manifest = FALSE,
method = 'gdc-client', ## use gdc-client tool to download data
directory = rnadir)
### Download miRNAs data
gdcRNADownload(project.id = 'TCGA-CHOL',
data.type = 'miRNAs',
write.manifest = FALSE,
method = 'gdc-client', ## use gdc-client tool to download data
directory = mirdir)
3.2 Data organization
### Parse RNAseq metadata
metaMatrix.RNA <- gdcParseMetadata(project.id = 'TCGA-CHOL',
data.type = 'RNAseq',
write.meta = FALSE)
# Filter duplicated samples in RNAseq metadata
metaMatrix.RNA <- gdcFilterDuplicate(metaMatrix.RNA)
# Filter non-Primary Tumor and non-Solid Tissue Normal samples in RNAseq metadata
metaMatrix.RNA <- gdcFilterSampleType(metaMatrix.RNA)
### Parse miRNAs metadata
metaMatrix.MIR <- gdcParseMetadata(project.id = 'TCGA-CHOL',
data.type = 'miRNAs',
write.meta = FALSE)
# Filter duplicated samples in miRNAs metadata
metaMatrix.MIR <- gdcFilterDuplicate(metaMatrix.MIR)
# Filter non-Primary Tumor and non-Solid Tissue Normal samples in miRNAs metadata
metaMatrix.MIR <- gdcFilterSampleType(metaMatrix.MIR)
### Merge raw counts data
# Merge RNAseq data
rnaCounts <- gdcRNAMerge(metadata = metaMatrix.RNA,
path = rnadir,
organized = FALSE, ## if target data are in folders
data.type = 'RNAseq')
# Merge miRNAs data
mirCounts <- gdcRNAMerge(metadata = metaMatrix.MIR,
path = mirdir,
organized = FALSE, ## if target data are in folders
data.type = 'miRNAs')
### TMM normalization and voom transformation
# Normalization of RNAseq data
rnaExpr <- gdcVoomNormalization(counts = rnaCounts, filter = FALSE)
# Normalization of miRNAs data
mirExpr <- gdcVoomNormalization(counts = mirCounts, filter = FALSE)
### Differential gene expression analysis
DEGAll <- gdcDEAnalysis(counts = rnaCounts,
group = metaMatrix.RNA$sample_type,
comparison = 'PrimaryTumor-SolidTissueNormal',
method = 'limma')
#data(DEGAll)
# All DEGs
deALL <- gdcDEReport(deg = DEGAll, gene.type = 'all')
# DE long-noncoding
deLNC <- gdcDEReport(deg = DEGAll, gene.type = 'long_non_coding')
# DE protein coding genes
dePC <- gdcDEReport(deg = DEGAll, gene.type = 'protein_coding')
Volcano plot and Heatmap
#Volcano plot
gdcVolcanoPlot(DEGAll)
# Barplot
gdcBarPlot(deg = deALL, angle = 45, data.type = 'RNAseq')
#Heatmap
#Heatmap is generated based on the heatmap.2() function in gplots package.
degName = rownames(deALL)
gdcHeatmap(deg.id = degName, metadata = metaMatrix.RNA, rna.expr = rnaExpr)
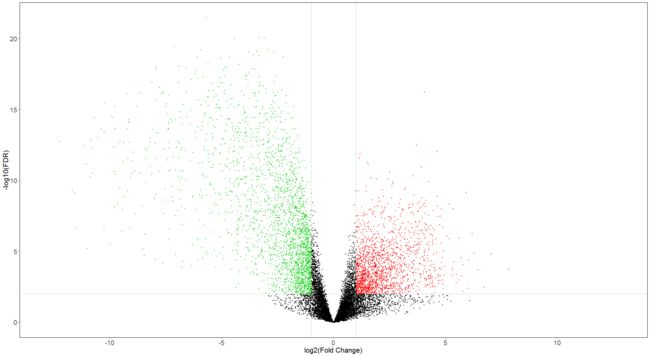
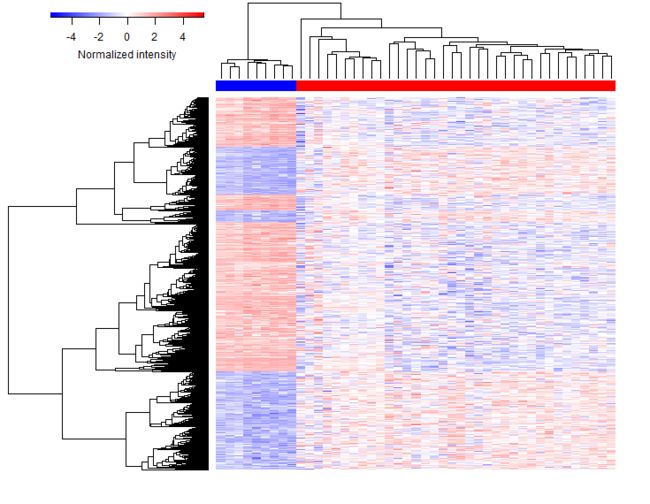
3.3 Competing endogenous RNAs network analysis
(ceRNAs network analysis)
### The 3 steps of ceRNAs network analysis:
# Hypergeometric test
# Pearson correlation analysis
# Regulation pattern analysis
### All of the 3 steps can be performed in a single function
### ceRNAs network analysis using internal databases
ceOutput <- gdcCEAnalysis(lnc = rownames(deLNC),
pc = rownames(dePC),
lnc.targets = 'starBase',
pc.targets = 'starBase',
rna.expr = rnaExpr,
mir.expr = mirExpr)
### ceRNAs network analysis using user-provided datasets
# load miRNA-lncRNA interactions
data(lncTarget)
lncTarget[1:3]
# load miRNA-mRNA interactions
data(pcTarget)
pcTarget[1:3]
ceOutput <- gdcCEAnalysis(lnc = rownames(deLNC),
pc = rownames(dePC),
lnc.targets = lncTarget,
pc.targets = pcTarget,
rna.expr = rnaExpr,
mir.expr = mirExpr)
### Network visulization in Cytoscape
# Filter potential ceRNA interactions
ceOutput2 <- ceOutput[ceOutput$hyperPValue<0.01 &
ceOutput$corPValue<0.01 & ceOutput$regSim != 0,]
# Edges and nodes can be simply imported into Cytoscape
# for network visualization
edges <- gdcExportNetwork(ceNetwork = ceOutput2, net = 'edges')
edges[1:5,]
nodes <- gdcExportNetwork(ceNetwork = ceOutput2, net = 'nodes')
nodes[1:5,]
write.table(edges, file='edges.txt', sep='\t', quote=F) ### Network of Cytoscape
write.table(nodes, file='nodes.txt', sep='\t', quote=F) ### Table of Cytoscape
### Correlation plot on a local webpage
shinyCorPlot(gene1 = rownames(deLNC),
gene2 = rownames(dePC),
rna.expr = rnaExpr,
metadata = metaMatrix.RNA)

3.4 Other downstream analyses
Univariate survival analysis
# CoxPH analysis
survOutput <- gdcSurvivalAnalysis(gene = rownames(deALL),
method = 'coxph',
rna.expr = rnaExpr,
metadata = metaMatrix.RNA)
# KM analysis
survOutput <- gdcSurvivalAnalysis(gene = rownames(deALL),
method = 'KM',
rna.expr = rnaExpr,
metadata = metaMatrix.RNA,
sep = 'median')
# KM plot on a local webpage by shinyKMPlot
shinyKMPlot(gene = rownames(deALL), rna.expr = rnaExpr,
metadata = metaMatrix.RNA)
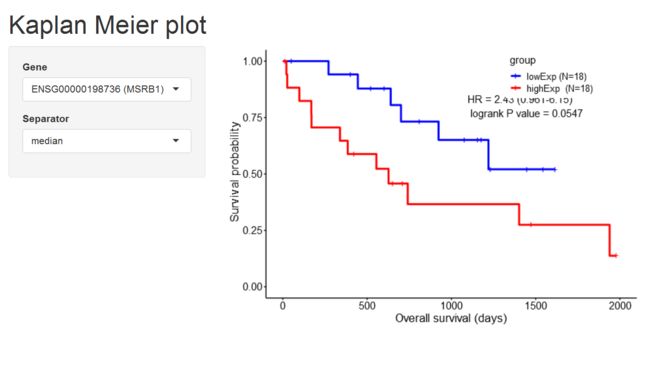
3.5 Functional enrichment analysis
All the functional enrichment analyses can be performed in a single function, including:
- Gene Ontology (BP, CC, MF) analysis
- KEGG pathway analysis
- Disease Ontology analysis
The speed was too slow and taked the top 100.
# Gene Ontology (BP, CC, MF) analysis #The speed is too slow and take the top 100.
enrichOutput <- gdcEnrichAnalysis(gene = rownames(deALL)[1:100], simplify = TRUE)
### This step may take a few minutes ###
# Step 1/5: BP analysis done!
# Step 2/5: CC analysis done!
# Step 3/5: MF analysis done!
# Step 4/5: KEGG analysis done!
# Step 5/5: DO analysis done!
#data(enrichOutput)
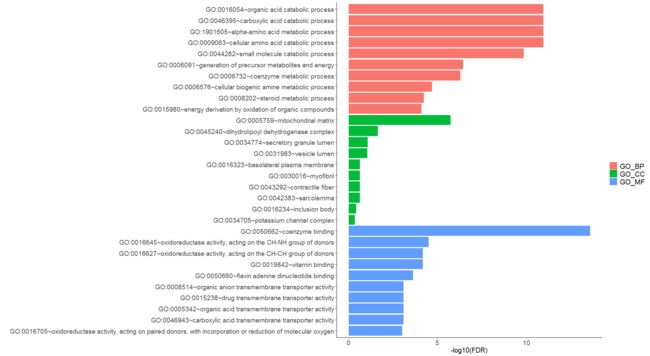
# Barplot
gdcEnrichPlot(enrichOutput, type = 'bar', category = 'GO', num.terms = 10)
write.csv(enrichOutput, "enrichOutput.csv")
# Bubble plot
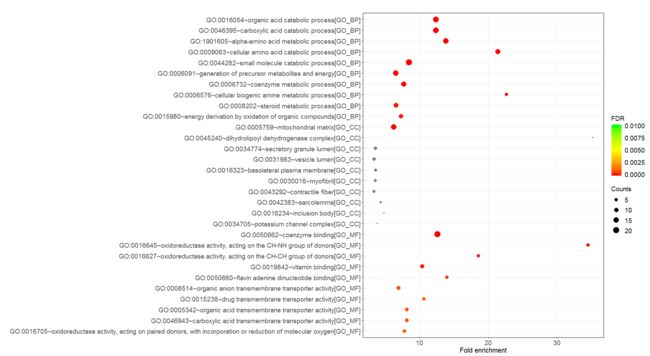
gdcEnrichPlot(enrichOutput, type='bubble', category='GO', num.terms = 10)
# KEGG pathway analysis
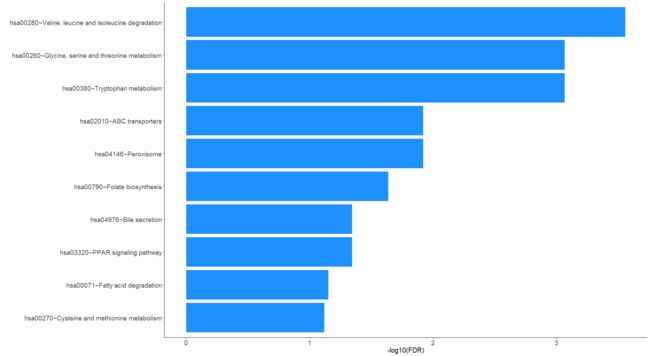
gdcEnrichPlot(enrichOutput, type = "bar", category = "KEGG", num.terms = 10, bar.color = "dodgerblue")
#bar.color = "chocolate1"
# Disease Ontology analysis
gdcEnrichPlot(enrichOutput, category='DO',type = 'bubble', num.terms = 20)
# View pathway maps on a local webpage
library(pathview)
deg <- deALL$logFC
names(deg) <- rownames(deALL)
pathways <- as.character(enrichOutput$Terms[enrichOutput$Category=='KEGG'])
shinyPathview(deg, pathways = pathways, directory = 'pathview')

View pathway maps报错如下:
Listening on http://127.0.0.1:6042
Warning: Error in %in%: object 'gene.idtype.bods' not found
[No stack trace available]
暂时没有找到解决办法
参考:
GDCRNATools的安装与使用---TCGA数据下载与分析工具
TCGA数据下载和整理工具----GDCRNATools
GDCRNATools.workflow.R
生信技能树公益视频合辑:学习顺序是linux,r,软件安装,geo,小技巧,ngs组学!
B站链接
YouTube链接
生信工程师入门最佳指南
学徒培养
生信技能树 -