R语言练习题
http://www.bio-info-trainee.com/3793.html
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
#######安装R包就不具体写了吧很久很久之前就装过了
1.打开 Rstudio 告诉我它的工作目录。 告诉我在你打开的Rstudio里面getwd()代码运行后返回的是什么
getwd()
[1] "C:/Users/Lenovo/Desktop/jns"
2.新建6个向量,基于不同的原子类型(重点是字符串,数值,逻辑值)
a<-1:5
class(a)
[1] "integer"
a
[1] 1 2 3 4 5
b<-c('snps','dna','rna')
b
[1] "snps" "dna" "rna"
class(b)
[1] "character"
c<-c(T,F,F,F,T,T,T,F,F,T,T)
c
[1] TRUE FALSE FALSE FALSE TRUE TRUE TRUE FALSE FALSE TRUE TRUE
class(c)
[1] "logical"
d<-seq(from =0, to = 30, by =5)
d
[1] 0 5 10 15 20 25 30
class(d)
[1] "numeric"
e<- c(15,16,19.99)
e
[1] 15.00 16.00 19.99
15,16本来没有小数点的,现在也出现小数位了
class(e)
[1] "numeric"
3.新建一些数据结构,比如矩阵,数组,数据框,列表等。重点是数据框,矩阵)
数组
ar<-array(1:10)
ar
[1] 1 2 3 4 5 6 7 8 9 10
矩阵
mat<- matrix(1:30,5)
mat
[,1] [,2] [,3] [,4] [,5] [,6]
[1,] 1 6 11 16 21 26
[2,] 2 7 12 17 22 27
[3,] 3 8 13 18 23 28
[4,] 4 9 14 19 24 29
[5,] 5 10 15 20 25 30
数据框
g<-paste0('gene',1:10)
gname <- c('a','b','c','d','e','f','g','h','i','j')
a1 <- c(floor(rnorm(5,5000,300)))
a2 <- c(floor(rnorm(5,3000,500)))
expr <- data.frame(id=g,genename=gname,s1=a1,s2=a2)
expr
id genename s1 s2
1 gene1 a 5136 2882
2 gene2 b 4418 3230
3 gene3 c 4789 3210
4 gene4 d 4603 2327
5 gene5 e 5602 2909
6 gene6 f 5136 2882
7 gene7 g 4418 3230
8 gene8 h 4789 3210
9 gene9 i 4603 2327
10 gene10 j 5602 2909
dim(expr)
[1] 10 4 ###(行/列)
View(expr)
4.在你新建的数据框进行切片操作,比如首先取第1,3行, 然后取第1, 3列
行
exprf<- expr[c(1,3), ]
exprf
id genename s1 s2
1 gene1 a 5136 2882
3 gene3 c 4789 3210
列
exprc<- expr[ , c(1,3)]
exprc
id s1
1 gene1 5136
2 gene2 4418
3 gene3 4789
4 gene4 4603
5 gene5 5602
6 gene6 5136
7 gene7 4418
8 gene8 4789
9 gene9 4603
10 gene10 5602
5.使用data函数来加载R内置数据集 rivers 描述它。并且可以查看更多的R语言内置的数据集:[https://mp.weixin.qq.com/s/dZPbCXccTzuj0KkOL7R31g]
data("rivers")
rivers
[1] 735 320 325 392 524 450 1459 135 465 600 330 336 280
[14] 315 870 906 202 329 290 1000 600 505 1450 840 1243 890
[27] 350 407 286 280 525 720 390 250 327 230 265 850 210
[40] 630 260 230 360 730 600 306 390 420 291 710 340 217
[53] 281 352 259 250 470 680 570 350 300 560 900 625 332
[66] 2348 1171 3710 2315 2533 780 280 410 460 260 255 431 350
[79] 760 618 338 981 1306 500 696 605 250 411 1054 735 233
[92] 435 490 310 460 383 375 1270 545 445 1885 380 300 380
[105] 377 425 276 210 800 420 350 360 538 1100 1205 314 237
[118] 610 360 540 1038 424 310 300 444 301 268 620 215 652
[131] 900 525 246 360 529 500 720 270 430 671 1770
length(rivers) ##多少个数据
[1] 141
head(rivers) ###查看前六个
[1] 735 320 325 392 524 450
class(rivers)
[1] "numeric"
min(rivers) ###最小值
[1] 135
srivers <-sort(rivers) ###排序
srivers
[1] 135 202 210 210 215 217 230 230 233 237 246 250 250
[14] 250 255 259 260 260 265 268 270 276 280 280 280 281
[27] 286 290 291 300 300 300 301 306 310 310 314 315 320
[40] 325 327 329 330 332 336 338 340 350 350 350 350 352
[53] 360 360 360 360 375 377 380 380 383 390 390 392 407
[66] 410 411 420 420 424 425 430 431 435 444 445 450 460
[79] 460 465 470 490 500 500 505 524 525 525 529 538 540
[92] 545 560 570 600 600 600 605 610 618 620 625 630 652
[105] 671 680 696 710 720 720 730 735 735 760 780 800 840
[118] 850 870 890 900 900 906 981 1000 1038 1054 1100 1171 1205
[131] 1243 1270 1306 1450 1459 1770 1885 2315 2348 2533 3710
6.下载 https://www.ncbi.nlm.nih.gov/sra?term=SRP133642 里面的 RunInfo Table 文件读入到R里面,了解这个数据框,多少列,每一列都是什么属性的元素。(参考B站生信小技巧获取runinfo table) 这是一个单细胞转录组项目的数据,共768个细胞,如果你找不到RunInfo Table 文件,可以点击下载,然后读入你的R里面也可以。
(1)输入网址
(2)点击Send results to Run selector
(3)Download---runinfo table
导入数据
srp <- read.table("SraRunTable.txt", header = T, sep = "\t")
View(srp)
这里出来的数据太多了,我就选了一小节
str(srp)
'data.frame': 768 obs. of 31 variables:
Experiment : chr "SRX3749902" "SRX3749903" "SRX3749904" "SRX3749905" ...
MBytes : int 8 8 4 4 5 4 9 3 6 8 ...
SRA_Sample : chr "SRS3006138" "SRS3006148" "SRS3006137" "SRS3006136" ...
Assay_Type : chr "RNA-Seq" "RNA-Seq" "RNA-Seq" "RNA-Seq" ...
$ AssemblyName : chr "GCF_000001635.20" "GCF_000001635.20" "GCF_000001635.20" "GCF_000001635.20"
dim(srp)
[1] 768 31
colnames(srp)
[1] "BioSample" "Experiment" "MBases"
[4] "MBytes" "Run" "SRA_Sample"
[7] "Sample_Name" "Assay_Type" "AssemblyName"
[10] "AvgSpotLen" "BioProject" "Center_Name"
[13] "Consent" "DATASTORE_filetype" "DATASTORE_provider"
[16] "InsertSize" "Instrument" "LibraryLayout"
[19] "LibrarySelection" "LibrarySource" "LoadDate"
[22] "Organism" "Platform" "ReleaseDate"
[25] "SRA_Study" "age" "cell_type"
[28] "marker_genes" "source_name" "strain"
[31] "tissue"
library("magrittr", lib.loc="D:/R/R-3.5.3/library")
library("dplyr", lib.loc="D:/R/R-3.5.3/library")
查看每一列的属性
for (i in colnames(srp)) paste(i,class(srp[,i])) %>% print()
[1] "BioSample character"
[1] "Experiment character"
[1] "MBases integer"
[1] "MBytes integer"
[1] "Run character"
[1] "SRA_Sample character"
[1] "Sample_Name character"
[1] "Assay_Type character"
[1] "AssemblyName character"
[1] "AvgSpotLen integer"
[1] "BioProject character"
[1] "Center_Name character"
[1] "Consent character"
[1] "DATASTORE_filetype character"
[1] "DATASTORE_provider character"
[1] "InsertSize integer"
[1] "Instrument character"
[1] "LibraryLayout character"
[1] "LibrarySelection character"
[1] "LibrarySource character"
[1] "LoadDate character"
[1] "Organism character"
[1] "Platform character"
[1] "ReleaseDate character"
[1] "SRA_Study character"
[1] "age character"
[1] "cell_type character"
[1] "marker_genes character"
[1] "source_name character"
[1] "strain character"
[1] "tissue character"
7.下载 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE111229 里面的样本信息sample.csv读入到R里面,了解这个数据框,多少列,每一列都是什么属性的元素。(参考 https://mp.weixin.qq.com/s/fbHMNXOdwiQX5BAlci8brA 获取样本信息sample.csv)如果你实在是找不到样本信息文件sample.csv,也可以点击下载。
sam<- read.csv(file = "sample.csv")
dim(sam)
[1] 768 12
ncol(sam)
[1] 12
View(sam)
for (i in colnames(sam)) paste(i,class(sam[,i])) %>% print()
[1] "Accession character"
[1] "Title character"
[1] "Sample.Type character"
[1] "Taxonomy character"
[1] "Channels integer"
[1] "Platform character"
[1] "Series character"
[1] "Supplementary.Types character"
[1] "Supplementary.Links character"
[1] "SRA.Accession character"
[1] "Contact character"
[1] "Release.Date character"
8.把前面两个步骤的两个表(RunInfo Table 文件,样本信息sample.csv)关联起来,使用merge函数(这道题有点迷啊)
打开两个数据看了一下发现sample.csv的Accession和RunInfo Table的Sample_Name 一样,那就用这两列合并吧
m <- merge(srp, sam, by.x = 'Sample_Name',by.y = 'Accession')
dim(m)
[1] 768 42
################我是一个去看了毕业晚会的分割线####################
################来自于收到了生姜洗发水的快乐####################
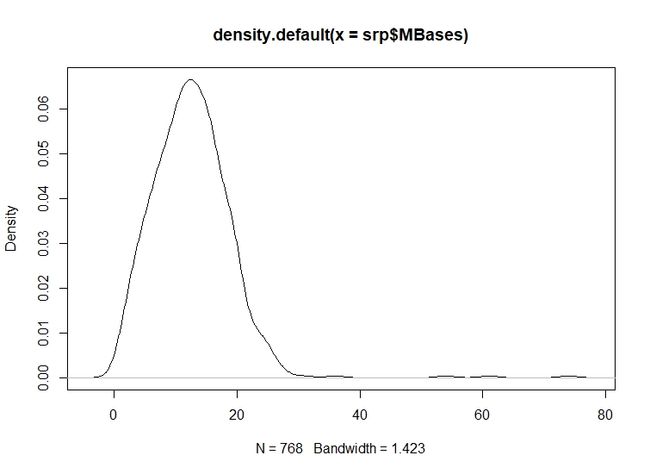
9.对前面读取的 RunInfo Table 文件在R里面探索其MBases列,包括 箱线图(boxplot)和五分位数(fivenum),还有频数图(hist),以及密度图(density) 。
boxplot(srp$MBases) ###箱线图

fivenum (srpMBases) ####频数图
density(srpMBases)
Data: srp$MBases (768 obs.); Bandwidth 'bw' = 1.423
x y
Min. :-4.269 Min. :0.0000000
1st Qu.:16.366 1st Qu.:0.0000353
Median :37.000 Median :0.0003001
Mean :37.000 Mean :0.0121039
3rd Qu.:57.634 3rd Qu.:0.0142453
Max. :78.269 Max. :0.0665647

10.前面读取的样本信息表格的样本名字根据下划线分割看第3列元素的统计情况。第三列代表该样本所在的plate。根据plate把关联到的 RunInfo Table 信息的MBases列分组检验是否有统计学显著的差异
m2<-m[, c("Title", "MBases")]
plate = unlist(lapply(m2[,1], function(x){
x
strsplit(x, "_")[[1]][3]
}))
strsplit函数字符串的分割函数,可以指定分割符,生成一个list
table(plate)
plate
0048 0049
384 384
t.test(m2[,2] ~ plate)####t检验
Welch Two Sample t-test
data: m2[, 2] by plate
t = 2.3019, df = 728.18, p-value = 0.02162 ###p<0.05
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
0.1574805 1.9831445
sample estimates:
mean in group 0048 mean in group 0049
13.08854 12.01823
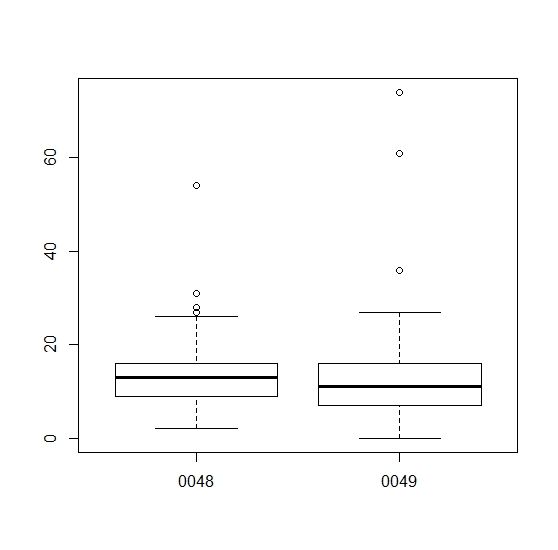
11.分组绘制箱线图(boxplot),频数图(hist),以及密度图(density) 。
boxplot(m2[,2] ~ plate)
 5.jpeg
5.jpeg
12.使用ggplot2把上面的图进行重新绘制。
library(ggplot2)
m3<- m2%>% mutate(plate = plate) ###m3比m2多一列plate
ggplot(m3, aes(x=m3MBases, color = plate)) + geom_boxplot()
ggplot(m3, aes(x = m3plate)) +geom_histogram()
有个小报错?但是不影响图stat_bin() using bins = 30. Pick better value withbinwidth
ggplot(m3, aes(x = m3plate)) +geom_density()



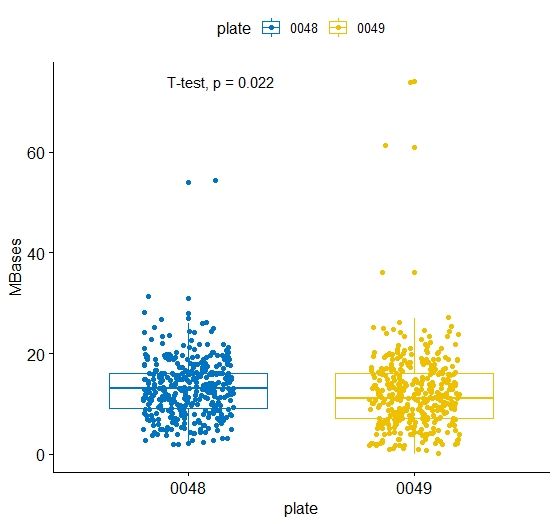
- 使用ggpubr把上面的图进行重新绘制。
library("ggpubr", lib.loc="D:/R/R-3.5.3/library")
p <- ggboxplot(m3, x = "plate", y = "MBases",
color = "plate", palette = "jco",
add = "jitter")
p
p + stat_compare_means(method = 't.test')
就会多出t检验的结果

- 随机取384个MBases信息,跟前面的两个plate的信息组合成新的数据框,第一列是分组,第二列是MBases,总共是384*3行数据。
ind<-sample(nrow (m3), 384)
data<-m3[indexes, ]
data2<-data[,c(3,1,2)]
dim(data2)
