从获取下机数据做质控、比对,到calling snp获得vcf文件这一步,网上已经有非常详细的教程,有GATK4.0和全基因组数据分析实践(上),还有Samtools+bcftools Call SNP等等。但是在群体遗传的研究中,我们经常需要比较不同群体的vcf文件,鉴定不同群体间基因组水平上发生分化的区域,并注释其发生分化的基因,网上对于这类群体选择信号分析的分享还比较少。
我通过计算Fst和TajimaD这两个经典的指数,来进行选择信号的分析。话不多说,先走一遍流程吧!
1、工具和文件的准备
工具:vcftools;snpEff;
文件:突变型群体的vcf文件(DA.vcf);包含了突变型和野生型两个群体的vcf文件(all.vcf);突变型群体信息(DA.txt);野生型群体信息(non_DA.txt);参考基因组的GFF注释文件(如果GFF注释文件中没有对应的GO编号信息,则还需要该物种基因ID与GO编号的对应信息文件annot.go.tab)
2、滑窗计算Fst和TajimaD值
vcftools可以通过设置窗口大小来计算染色体(或scaffolds)上指定区域的Fst和TajimaD的值,但不足的是在计算TajimaD值时,不能设置步长(可使用VCF-kit进行可设置步长的计算),因此,为了方便后续的分析,我在这里计算Fst和TajimaD时,只设置了窗口大小(10000),未设置步长。
vcftools --vcf all.vcf --weir-fst-pop DA.txt --weir-fst-pop non_DA.txt --fst-window-size 10000 --out da_nonda
生成da_nonda.windowed.weir.fst 文件
CHROM BIN_START BIN_END N_VARIANTS WEIGHTED_FST MEAN_FST
1 1 10000 7 0.0976319 0.0971726
1 10001 20000 61 0.0511299 0.0499982
1 20001 30000 49 0.0372642 0.0402562
1 30001 40000 49 0.0679565 0.073192
1 40001 50000 17 0.0299695 0.0355619
1 50001 60000 56 0.101204 0.105008
1 60001 70000 180 0.0951801 0.100651
1 70001 80000 21 0.07005 0.0728101
1 80001 90000 60 0.170809 0.178863
1 90001 100000 21 0.0920029 0.10329
vcftools --vcf DA.vcf --TajimaD 10000 --out DA
生成DA.Tajima.D文件
CHROM BIN_START N_SNPS TajimaD
1 0 9 2.88793
1 10000 40 3.12075
1 20000 70 2.90904
1 30000 90 3.37423
1 40000 49 3.24297
1 50000 88 3.31503
1 60000 329 3.52148
1 70000 26 2.38513
1 80000 82 2.30261
1 90000 82 2.37531
1 100000 12 2.48976
1 110000 86 2.43792
1 120000 52 2.95367
1 130000 57 3.03159
1 140000 71 3.06152
1 150000 142 3.478
1 160000 0 nan
1 170000 2 0.34569
1 180000 0 nan
3、对Fst和TajimaD值进行筛选、排序
首先对生成Fst文件进行排序,将文件按照第六列,也就是MEAN_FST这列值,从大到小进行排序
sort -nr -k6 da_nonda.windowed.weir.fst
1 12910001 12920000 1 2.47818e-17 2.47818e-17
11 11750001 11760000 1 2.47818e-17 2.47818e-17
11 3990001 4000000 2 2.18381e-18 2.29795e-18
10 7260001 7270000 1 2.10168e-17 2.10168e-17
7 16280001 16290000 2 1.0842e-17 1.23909e-17
1 13520001 13530000 2 1.0842e-17 1.23909e-17
7 23470001 23480000 1 0.9375 0.9375
8 18430001 18440000 1 0.931034 0.931034
1 8430001 8440000 1 0.845361 0.845361
9 9550001 9560000 1 0.833333 0.833333
9 5930001 5940000 1 0.833333 0.833333
9 13450001 13460000 1 0.833333 0.833333
排序之后发现,由于有些值过小,在使用科学计数法时排在了正常计数值的前面,为了去掉这个错误,先把使用科学计数法的值,统一归为0,再进行排序
awk '{if($6~/e/)$6=0}1' da_nonda.windowed.weir.fst > da_nonda.window.clean.fst && sort -rn -k6 da_nonda.windowed.clean.fst > da_nonda.windowed.sort.fst
对文件第2列做减1处理,方便后面与TajimaD匹配
awk '{$2 = $2-1;print$0}' sp1_da.window.sort.fst > sp1_da.sort.fst
统计da_nonda.sort.fst有多少行
wc -l da_nonda.sort.fst
17824
取Fst值最大的前10%窗口,作为候选选择区域
head -n 1782 da_nonda.sort.fst > da_nonda.top0.1.fst
对于TajimaD文件,先清除掉第4列为nan的行,再对其进行排序,取前5%和后5%的值
sed -e '/nan/d' DA.Tajima.D > DA.clean.tajimaD && sort -nr -k4 DA.clean.tajimaD > DA.sort.tajimaD
wc -l DA.sort.tajimaD
19484
head -n 974 DA.sort.tajimaD > DA.top0.05.tajimaD
tail -n 974 DA.sort.tajimaD > DA.bot0.05.tajimaD
取Fst前10%区域和TajimaD前5%(后5%)区域的交集,并按照染色体(scaffolds)顺序排序,将得到的这些窗口作为受选择区域
cat DA.top0.05.tajimaD da_nonda.top0.1.fst > DA.top && awk 'pass==1 { count[$1,$2]++ } pass==2 { if(count[$1,$2]>1) print }' pass=1 DA.top pass=2 DA.top > DA.top.site
wc -l DA.top.site
40
head -n 20 DA.top.site > DA.top.keep.site
sort -n -k1 DA.top.keep.site > DA.top.sort.site
得到的文件如下
1 17540000 2 1.9458
1 28520000 3 1.96716
2 17140000 5 2.41094
2 19520000 2 2.03336
2 19620000 2 1.97256
2 19630000 2 2.03336
2 21790000 2 1.92526
2 28710000 2 2.03336
3 12170000 12 2.63244
3 14190000 12 2.74771
3 25930000 4 1.94295
3 4700000 4 2.25325
4 8030000 5 2.04905
5 8680000 16 2.6894
6 13390000 7 2.06337
6 22180000 3 2.0041
7 23620000 2 1.92526
8 8650000 3 2.24976
11 5080000 3 2.04485
12 1160000 6 2.23514
4、使用snpEff对DA.vcf文件进行注释
snpEff的教程很多,先SnpEff 配置基因组注释文件,再snpEFF注释vcf-笔记,最终得到包含注释信息的DA.annotation.vcf
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT sample12 sample13 sample14 sample15 sample16 sample17 samp
le18 sample19 sample20 sample21
1 3923 . T C 98 PASS BQB=0.169762;HOB=0.5;ICB=1;MQ=40;MQ0F=0.0322581;MQB=0.000633889;MQSB=0.975265;RPB=0.103031;SGB=-0.662043;VDB=0.07878
29;DP=198;DP4=60,33,30,28;AN=10;AC=5;ANN=C|intergenic_region|MODIFIER|CHR_START-VJ01Gene00001|CHR_START-VJ01Gene00001|intergenic_region|CHR_START-VJ01Gene00001|||n.3923T>C|
||||| GT:AD:ADF:ADR:DP:PL:SP ./.:.:.:.:.:.:. 0/1:12,9:8,6:4,3:21:55,0,255:0 0/1:27,14:17,8:10,6:41:132,0,255:1 ./.:.:.:.:.:.:. 0/1:15,11:9,6:6,5:28:108,0,255:0
0/1:18,13:11,5:7,8:31:105,0,255:5 ./.:.:.:.:.:.:. 0/1:21,9:15,4:6,5:30:70,0,255:6 ./.:.:.:.:.:.:. ./.:.:.:.:.:.:.
5、从物种的基因ID到GO ID
得到分化区域内SNP的注释信息,并去掉intron区和同义突变的位点
awk '$3 = "vcftools --vcf DA.annotation.vcf --chr"" " $1" " "--from-bp"" "$2" " "--to-bp"" "$2+10000" " "--recode --recode-INFO-all --out"" "$1"_"$2 {print $3}' DA.top.sort.site > vcf.sh
sh vcf.sh
grep -v "int" *.recode.vcf|grep -v "synonymous_variant" > non_int_synon.vcf
提取SNP位点所对应的基因ID信息
awk '{print $8}' non_int_synon.vcf|grep -o 'VJ01Gene[0-9][0-9][0-9][0-9][0-9]' > geneID.txt
去重复
sort -n geneID.txt | uniq > clean_geneID.txt
将物种的基因ID与GO数据库的ID对应
awk 'ARGIND==1{a[$1]=$0} ARGIND==2 && ($1 in a) {print $0}' clean_geneID.txt annot.go.tab > geneID_goID.txt
提取GO ID
awk '{print $2}' geneID_goID.txt > goID.txt
less goID.txt
GO:0004725
GO:0006570
GO:0035335
GO:0004190
GO:0006508
GO:0004190
GO:0005764
GO:0006508
这样就得到了Fst top0.1和TajimaD top 0.05两者都存在的GO ID,即受到平衡选择的基因,TajimaD bot 0.05筛选的是受到定向选择的基因,做法也是一样的。
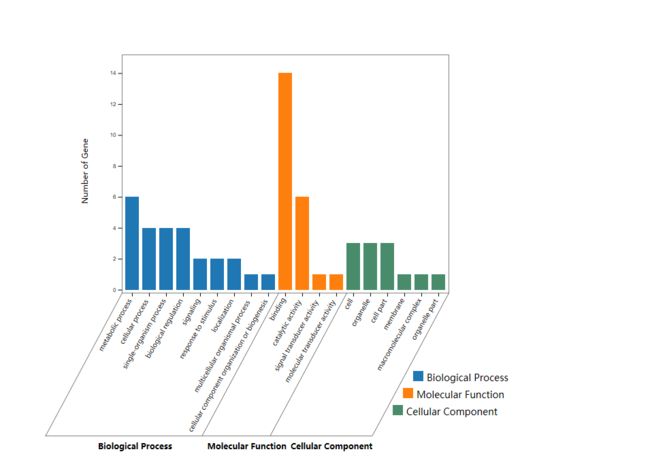
最后,对我们得到的基因进行GO富集分析,在线GO富集的工具有很多,基迪奥在线云分析平台和遗传所王秀杰课题组开发的GOEAST平台都很方便做。基迪奥平台的富集分析结果如下
写在最后:从去年11月拿到下机的重测序数据,并简单搭建了服务器分析环境,一路摸着石头过河,战战兢兢,在度娘和github两位老师的指导下才逐步入门,希望和更多做群体重测序的小伙伴交流啊!QQ:657231499