本文重复《甜过初恋!这次是真的批量做TCGA的生存分析》, 作者果子大神。
地址: https://mp.weixin.qq.com/s/fuB0bLyllL_33_Shz7rKnw
1. 安装包
# Load the bioconductor installer.
source("https://bioconductor.org/biocLite.R")
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
# Install the main RTCGA package
biocLite("RTCGA")
# Install the clinical and mRNA gene expression data packages
biocLite("RTCGA.clinical")
biocLite("RTCGA.mRNA")
- 加载包
library(RTCGA)
#了解数据
infoTCGA <- infoTCGA()
# Create the clinical data
library(RTCGA.clinical)
clin <- survivalTCGA(BRCA.clinical) #到这里临床部分的信息已经获得啦
-
看下数据类型和内容
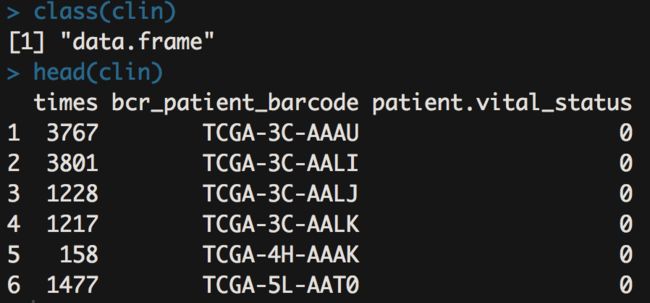 image.png
image.png
2. 获得gene表达数据
library(RTCGA.mRNA) #加载数据包
class(BRCA.mRNA) #查看数据类型发现是个数据框
dim(BRCA.mRNA) #看一下数据维度发现有590个样本,17815个基因
BRCA.mRNA[1:5, 1:5] #看一下部分数据样子
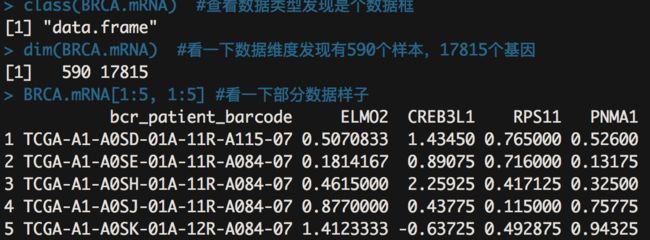
image.png
- 挑选几个基因的表达数据出来,融合到生存的数据上去
- 我们使用管道符号%>%,他的作用是 把前一个计算得到结果,作为第二个函数的参数
library(dplyr)
BRCA.mRNA %>%
# then make it a tibble (nice printing while debugging)
as_tibble() %>%
# then get just a few genes,这里是测试用
dplyr::select(bcr_patient_barcode, PAX8, GATA3, ESR1) %>%
# then trim the barcode (see head(clin), and substr)
mutate(bcr_patient_barcode = substr(bcr_patient_barcode, 1, 12)) %>%
# then join back to clinical data
inner_join(clin, by="bcr_patient_barcode") %>% exprSet
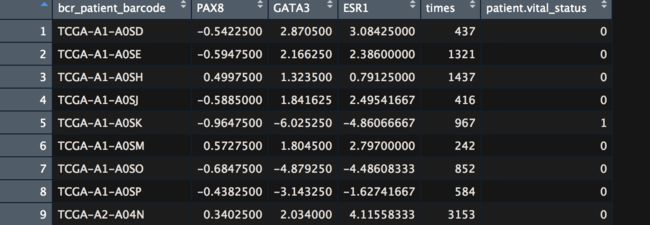
image.png
3. 开始做生存分析
library(survival)
library(ggplot2)
library(ggpubr)
library(magrittr)
library(survminer)
- 对需要做生存分析的样本分组,把连续变量变成分类变量,这里选择测试的基因是GATA3
group <- ifelse(exprSet$GATA3>median(exprSet$GATA3),'high','low')
- 构建生存对象,并且进行数据处理
sfit <- survfit(Surv(times, patient.vital_status)~group, data=exprSet)
sfit
summary(sfit)
ggsurvplot(sfit, conf.int=F, pval=TRUE)

image.png
4. 批量数据做生存分析
- 获得完整的表达量数据
library(dplyr)
exprSet <- BRCA.mRNA %>%
# then make it a tibble (nice printing while debugging)
as_tibble() %>%
# then trim the barcode (see head(clin), and ?substr)
mutate(bcr_patient_barcode = substr(bcr_patient_barcode, 1, 12)) %>%
# then join back to clinical data
inner_join(clin, by="bcr_patient_barcode")
- 构建生存对象my.surv
library(survival)
my.surv <- Surv(exprSet$times, exprSet$patient.vital_status)
- 尝试使用并行运算提速
library(parallel)
#detectCores()检查当前电脑可用核数
cl.cores <- detectCores()
#makeCluster(cl.cores)使用刚才检测的核并行运算,我的电脑是8核
cl <- makeCluster(8)
#这是坑,parApply里面用到的函数以及变量都需要申明,不声明就必须用模块
clusterExport(cl,c("exprSet","my.surv"))
#length(names(esprSet))-2,为什么减去2,因为之前小规模测试时,我们知道最后两个是time和event,不是表达量
#数据从25开始,原因是从2开始会报错,暂时无法解决,还有要注意是parApply,A要大写的
log_rank_p <- parApply(cl,exprSet[,25:length(names(exprSet))-2],2,function(values){
group=ifelse(values>median(na.omit(values)),'high','low')
kmfit2 <- survival::survfit(my.surv~group,data=exprSet)
#plot(kmfit2)
data.survdiff=survival::survdiff(my.surv~group)
p.val = 1 - pchisq(data.survdiff$chisq, length(data.survdiff$n) - 1)
})
stopCluster(cl)
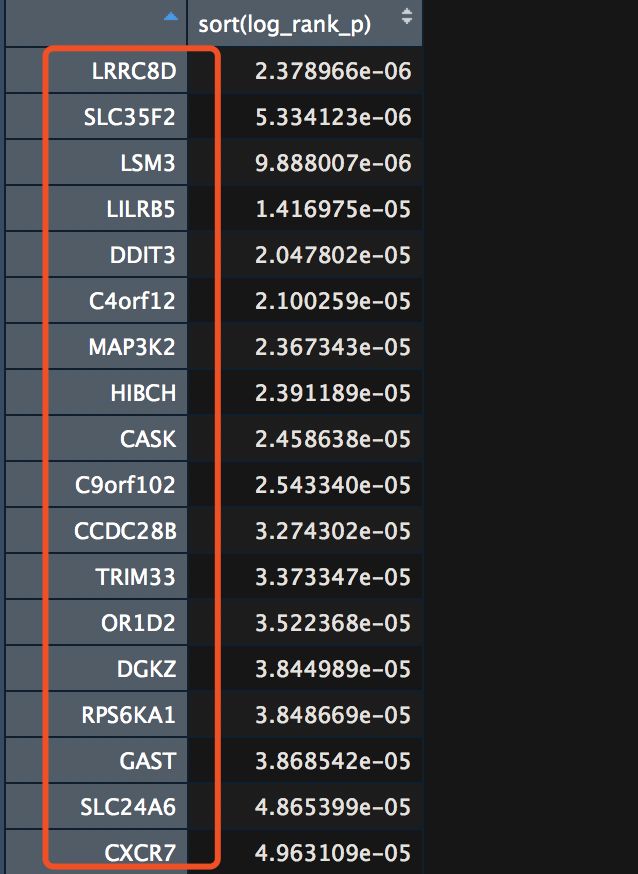
- 找出小于0.05的P.val
log_rank_p <- log_rank_p[log_rank_p<0.05]
- 筛选后排序,并获得基因名
gene_diff <- as.data.frame(sort(log_rank_p))
找到2153个gene
- 保存与读取数据
save(gene_diff,file = "gene_df.Rda")
load(file = "gene_df.Rda")
- 找个基因看看效果
library(survminer)
group <- ifelse(exprSet$MAP3K2>median(exprSet$MAP3K2),'high','low')
sfit <- survfit(Surv(times, patient.vital_status)~group, data=exprSet)
ggsurvplot(sfit, conf.int=FALSE, pval=TRUE)
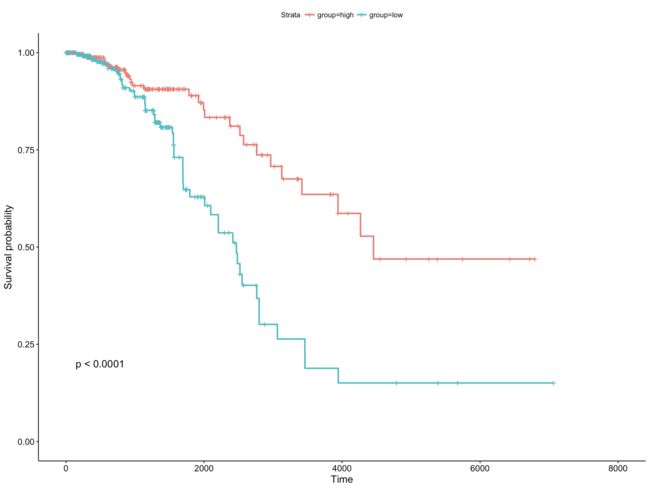
image.png
-
这些基因想看哪个就看哪个