参考:
1.第六章 scRNA-seq数据分析
2.初生牛犊--模仿cell research (IF 17+) 文章的一张图
3.Seurat包学习笔记(一):Guided Clustering Tutorial
4.2019-10-22 R语言Seurat包下游分析-1

目录
1.数据下载
2.数据读取
3.质量控制
3.1 载入注释
3.2 质控及其细胞选取
4.标准化
5.高差异基因 (HVGs)
6. 二次标准化
6.1 缩放的标准
6.2 删除不需要的数据
7. PCA 分析
8. 决定需要考虑的PC
9.细胞分集
10.降维后,用聚类的类别可视化【UMAP/tSNE】
11.寻找分集标记基因
12.对分集细胞进行标记
Seurat是一个常用的clustering的软件包。它的最大贡献在于将tSNE图引入到scRNA-seq cluter的图形化展示中来。tSNE是一种降维算法,它将多维的PCA分析降维到2维图形中,使用人们很方便的在二维图形中区分不同类型的细胞。
安装并加载所需的R包
- 目前最新版的Seurat包都是基于3.0以上的,可以直接通过install.packages命令进行安装。
# 设置R包安装镜像
options(repos="https://mirrors.tuna.tsinghua.edu.cn/CRAN/")
install.packages('Seurat')
library(Seurat)
library(dplyr)
library(patchwork)
# 查看Seurat包的版本信息
packageVersion("Seurat")
- 如果想安装早期2.0版本的Seurat包可以通过devtools包进行安装
# 先安装devtools包
install.packages('devtools')
# 使用devtools安装指定版本的Seurat包
devtools::install_version(package = 'Seurat', version = package_version('2.3.4'))
library(Seurat)
Seurat的原教程
1.数据下载
10X genomics 数据下载地址 : 可以在这里下载到
注释文件: ensembl数据库中下载 human gtf文件 。为下文计算每个细胞中不同类型的表达基因比例做装备。(如:编码基因,线粒体基因,长链非编码基因等等比例)
2.数据读取
Seurat教程中就是10X Genomics数据。通过Read10X来读取。

library(Seurat)
library(dplyr)
library(ggplot2)
# 读取Peripheral Blood Mononuclear Cells (PBMC)数据
# 解压文件应该是hg19文件夹,切换到这个目录中:
setwd("C:/pbmc3k_filtered_gene_bc_matrices/filtered_gene_bc_matrices/hg19")
# 直接用Read10X 读取文件。
pbmc.data <- Read10X(data.dir = "./")
pbmc是一个稀疏矩阵.用. 代替0,优化内存.
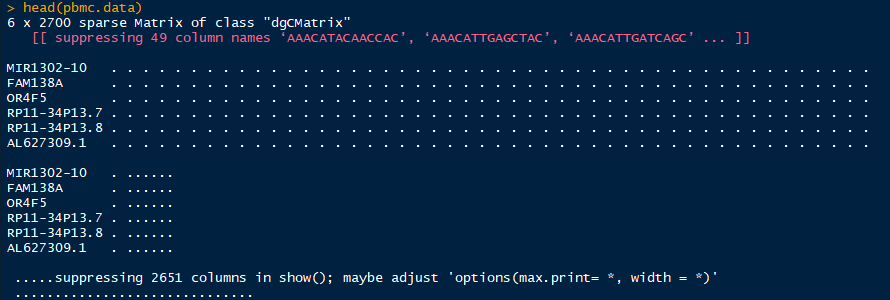
建立Seurat 对象
pbmc <- CreateSeuratObject(counts = pbmc.data, min.cells = 3,
min.features = 200, project = "10X_PBMC")
- counts = pbmc.data : 未标准化的数据,如原始计数或TPMs .
- min.cells =3 : 基因过滤指标, 基因至少在3个细胞中表达.
- min.feature =200 : 细胞过滤指标,一个细胞中表达基因至少有200.
- project = "10X_PBMC" : 设置项目名称,给每一个细胞打一个标签,都是来自10X_PBMC 项目
获得的pbmc 对象,有13714个基因,2700个细胞用过了初始的质控标准(min.cells =3;min.feature =200)
Tips: 对于拿到UMI计数表格的情况,我们只要能够够建好一个dgTMatrix就可以了。问题的关键是如何在有限的内存下构建dgTMatrix。可以采取分段读入的办法。
比如下面的方法:
library(Seurat)
library(Matrix)
nUMI.summary.file <- "data/umi_expression_matrix.tsv"
counts <- list()
con <- file(nUMI.summary.file, open="r")
## 读取文件头
header <- readLines(con, n=1, warn=FALSE)
header <- strsplit(header, "\\t")[[1]]
## 读取计数
i <- 1
while(length(buf <- readLines(con, n=1e6, warn=FALSE))>0){
buf <- as.matrix(read.delim(text=buf, header=FALSE, row.names=1))
counts[[i]] <- Matrix(buf, sparse=TRUE)
i <- i + 1
}
counts <- do.call(rbind, counts)
rownames(counts)[1:5]
colnames(counts) <- header
## 初始化一个Seurat对象
d <- CreateSeuratObject(counts = counts, min.cells = 10,
min.features = 200, project = "test_project")
3.质量控制
质量控制从一开始构建Seurat对象的时候就已经开始了。那个时候, 我们依据的是基因表达的细胞数以及细胞内被检测到的基因数来进行初步筛选的(min.cells =3;min.feature =200)。 Seurat还提供了更多的筛选手段,比如线粒体DNA含量等。 所以需要gtf注释文件,得到哪些基因是线粒体基因,哪些基因是编码基因.
3.1 载入注释
对于线粒体基因,通常为 MT-启始的,所以人们常常用这一特点来标定线粒体DNA。 但不是全部情形,下面是示例如何通过 ensembl GFF文件 进行读取文件。
library(rtracklayer)
# BiocManager::install("rtracklayer")
gtf <- import("Homo_sapiens.GRCh37.87.chr.gtf.gz")
gtf <- gtf[gtf$gene_name!=""]
gtf <- gtf[!is.na(gtf$gene_name)]
## protein coding
protein <-
gtf$gene_name[gtf$transcript_biotype %in%
c("IG_C_gene", "IG_D_gene", "IG_J_gene", "IG_LV_gene",
"IG_M_gene", "IG_V_gene", "IG_Z_gene",
"nonsense_mediated_decay", "nontranslating_CDS",
"non_stop_decay", "polymorphic_pseudogene",
"protein_coding", "TR_C_gene", "TR_D_gene", "TR_gene",
"TR_J_gene", "TR_V_gene")]
## mitochondrial genes
mito <- gtf$gene_name[as.character(seqnames(gtf)) %in% "MT"]
## long noncoding
lincRNA <-
gtf$gene_name[gtf$transcript_biotype %in%
c("3prime_overlapping_ncrna", "ambiguous_orf",
"antisense_RNA", "lincRNA", "ncrna_host", "non_coding",
"processed_transcript", "retained_intron",
"sense_intronic", "sense_overlapping")]
## short noncoding
sncRNA <-
gtf$gene_name[gtf$transcript_biotype %in%
c("miRNA", "miRNA_pseudogene", "misc_RNA",
"misc_RNA_pseudogene", "Mt_rRNA", "Mt_tRNA",
"Mt_tRNA_pseudogene", "ncRNA", "pre_miRNA",
"RNase_MRP_RNA", "RNase_P_RNA", "rRNA", "rRNA_pseudogene",
"scRNA_pseudogene", "snlRNA", "snoRNA",
"snRNA_pseudogene", "SRP_RNA", "tmRNA", "tRNA",
"tRNA_pseudogene", "ribozyme", "scaRNA", "sRNA")]
## pseudogene
pseudogene <-
gtf$gene_name[gtf$transcript_biotype %in%
c("disrupted_domain", "IG_C_pseudogene", "IG_J_pseudogene",
"IG_pseudogene", "IG_V_pseudogene", "processed_pseudogene",
"pseudogene", "transcribed_processed_pseudogene",
"transcribed_unprocessed_pseudogene",
"translated_processed_pseudogene",
"translated_unprocessed_pseudogene", "TR_J_pseudogene",
"TR_V_pseudogene", "unitary_pseudogene",
"unprocessed_pseudogene")]
annotations <- list(protein=unique(protein),
mito=unique(mito),
lincRNA=unique(lincRNA),
sncRNA=unique(sncRNA),
pseudogene=unique(pseudogene))
names(annotations )
annotations <- annotations[lengths(annotations)>0]
3.2 质控及其细胞选取
# Seurat会计算基因数以及UMI数 (nFeature and nCount).
# Seurat会将原始数据保存在RNA slot中,
# 每一行对应一个基因,每一列对应一个细胞.
slotNames(pbmc)
## [1] "assays" "meta.data" "active.assay" "active.ident" "graphs"
## [6] "neighbors" "reductions" "project.name" "misc" "version"
## [11] "commands" "tools"
names(pbmc@assays) ## Assays(pbmc)
## [1] "RNA"
head(Idents(pbmc)) ## 查看一下当前的细胞分组
## AAACATACAACCAC AAACATTGAGCTAC AAACATTGATCAGC AAACCGTGCTTCCG AAACCGTGTATGCG
## 10X_PBMC 10X_PBMC 10X_PBMC 10X_PBMC 10X_PBMC
## AAACGCACTGGTAC
## 10X_PBMC
## Levels: 10X_PBMC
下面将运用到注释文件,计算每一个细胞中各种类型基因,占所有基因比例。比如说编码基因比例0.85,线粒体基因比例0.10等等。
- PercentageFeatureSet : 函数就是计算各种基因所占比例
# 在计算比例时,使用目标基因中的数值除以总的数值。
# `[[`运算符可以给metadata加column.
percent <- lapply(annotations, function(.ele){
PercentageFeatureSet(pbmc, features = rownames(pbmc)[rownames(pbmc) %in% .ele])
})
# AddMetaData 会在[email protected]中加一列。这些信息都会在QC中使用到。
for(i in seq_along(percent)){
pbmc[[paste0("percent.", names(percent)[i])]] <- percent[[i]]
}
- 也可以手动对meta.data进行编辑. 添加一列对细胞进行标示(s_A,s_B)
id <- sample(c("s_A", "s_B"), nrow([email protected]), replace = TRUE)
names(id) <- rownames([email protected]) #必须有names
[email protected]$f1 <- id
注释后结果如下:
head([email protected])
## orig.ident nCount_RNA nFeature_RNA percent.protein percent.mito
## AAACATACAACCAC 10X_PBMC 2419 779 97.02356 3.0177759
## AAACATTGAGCTAC 10X_PBMC 4903 1352 96.45115 3.7935958
## AAACATTGATCAGC 10X_PBMC 3147 1129 93.83540 0.8897363
## AAACCGTGCTTCCG 10X_PBMC 2639 960 98.78742 1.7430845
## AAACCGTGTATGCG 10X_PBMC 980 521 96.53061 1.2244898
## AAACGCACTGGTAC 10X_PBMC 2163 781 91.77069 1.6643551
## percent.lincRNA percent.sncRNA percent.pseudogene f1
## AAACATACAACCAC 84.82844 0.20669698 0 s_B
## AAACATTGAGCTAC 86.33490 0.12237406 0 s_A
## AAACATTGATCAGC 84.30251 0.09532888 0 s_A
## AAACCGTGCTTCCG 75.74839 0.07578628 0 s_B
## AAACCGTGTATGCG 76.73469 0.30612245 0 s_B
## AAACGCACTGGTAC 86.54646 0.04623209 0 s_A
orig.ident 是细胞标示.初始化seurat 时候,orig.ident 为10x_pbmc , 当我们需要改变orig.ident时,我们需要改变的是ident slot.
# 试着改变分组。
olID <- Idents(pbmc)
Idents(pbmc) <- as.factor(id)
head(Idents(pbmc))
## AAACATACAACCAC AAACATTGAGCTAC AAACATTGATCAGC AAACCGTGCTTCCG AAACCGTGTATGCG
## s_B s_A s_A s_B s_B
## AAACGCACTGGTAC
## s_A
## Levels: s_A s_B
Idents(pbmc) <- olID ## 变回来
Tips : 上面是通过自己gtf中提取信息,进行注释,同时Seurat 里面也可以大体上按照特征进行标记.
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
可视化开始啦~
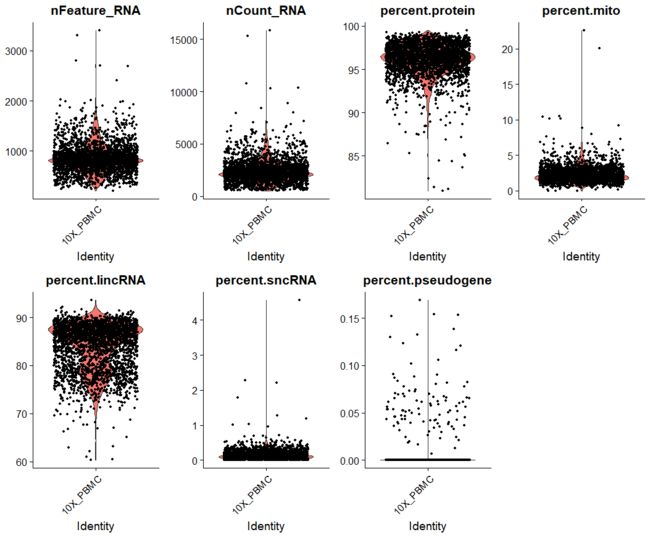
- 【一维可视化】对上面pbmc 所有注释的信息,进行可视化
VlnPlot(object = pbmc,
features = c("nFeature_RNA", "nCount_RNA",
paste0("percent.", names(percent))),
ncol = 4)
可以看到各种特征分布情况,进行设置特征的阈值,过滤细胞。
- nFeature_RNA :每个细胞中检测到的基因数目分布情况。
- nCount_RNA : 每个细胞中检测到的read 数目分布情况。
- percent.protein : 每个细胞中编码蛋白基因所占比例分布情况。
- percent.mito : 每个细胞中线粒体相关基因所占比例分布情况。
- ......
-
【二维可视化】选取两个特征进行画图 : FeatureScatter
# FeatureScatter是一个用来画点图的工具,可以比较不同的metadata。 plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mito") plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA") plot1 + plot2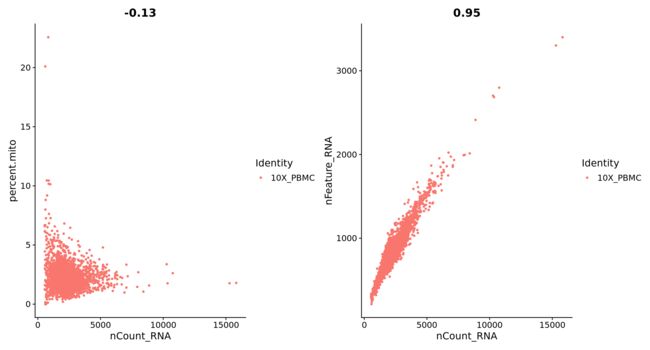 image.png
image.png
开始真正的细胞过滤啦~
从VlnPlot中,我们可以得到大多数细胞集中的范围,通过这些信息,可以设置过滤条件。
比如可以使用unique gene counts, percent.protein, percentage.mito来进行过滤。
这里我们设置了200 < nGene < 2500, 0.90 < percent.protein < Inf, -Inf < percent.mito < 0.05.
## 查看维度信息
dim(pbmc)
## [1] 13714 2700
## 开始过滤
pbmc <- subset(x = pbmc,
subset = nFeature_RNA > 200 & nFeature_RNA < 2500 &
percent.protein > 95 &
percent.mito < 5) ## something wrong
## 查看维度信息
dim(pbmc)
## [1] 13714 2142
4.标准化
标准化是一个比较简单的过程,使用的是"logNormalize", 就是将每个基因的表达量对该细胞总表达量进行平衡,然后乘以一个因子(scale factor, 默认值为10,000), 然后中进行对数转换。
pbmc <- NormalizeData(pbmc)
# 等同于
pbmc <- NormalizeData(seurat.obj,
normalization.method = "LogNormalize", scale.factor = 10000)
# 方法是LogNormalize
# scale.factor是它默认的
5.高差异基因 (HVGs)
在单细胞测序中,有些基因会在不同的细胞表现出高度的差异表达。Seurate可以对这些基因进行标记。
如果手动计算,相当于计算变异系数,而不是标准差。因为标准差对于数据呈现对称分布,及单峰情况体现差异比较好,但是对于双峰情况,变异系数cv 可能更好。
-
Tips:
使用
FindVariableFeatures完成差异分析,选择数据集中差异较高的特征基因(默认2000)并用于下游分析。 FindVariableFeatures 在2.0是FindVariablegene。包更新的很快.
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst",
nfeatures = 2000)
# 找到变化最大的十个基因
top10 <- head(VariableFeatures(pbmc), 10)
top10
## [1] "PPBP" "GNLY" "S100A9" "LYZ" "IGLL5" "PF4" "FTL" "FTH1"
## [9] "GNG11" "FCER1A"
- selection.method : 默认是“vst” ,也可以选择"roc"
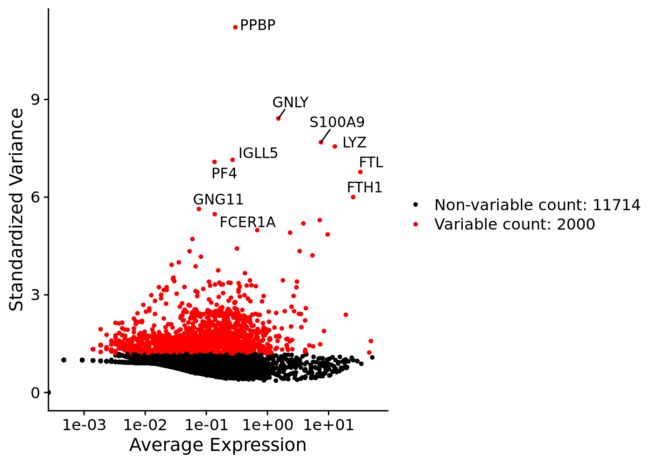
- nfeatures : 需要得到多少个高变异的基因。此处为2000个基因,下图可视化中标红的基因。
对HVGs 进行可视化
plot1 <- VariableFeaturePlot(pbmc)
LabelPoints(plot = plot1, points = top10, repel = TRUE)
- 红色的HVGs 有2000个,从上面 FindVariableFeatures ,命令参数就可以知道.
6. 二次标准化
从上图中,我们可以看到很多基因都表现出很大的差异性,但是这些差异性并不一定都是事实,有些可能是因为实验,或者数据采集的方式导入的。因此人们想尽量的消除这些因素带来的差异。这些差异有可能是因为batch-effect, 或者cell alignment rate,或者其它。这些因素都可以写入metadata中,然后通过ScaleData函数来消除。
ScaleData 可以通过metadata信息,消除很多其他因素影响。
6.1 缩放的标准
应用线性变换缩放数据,这是一个标准的预处理步骤,比PCA等降维技术更重要。使用ScaleData函数:
(1)使每个基因在所有细胞间的表达量均值为0
(2)使每个基因在所有细胞间的表达量方差为1
缩放操作给予下游分析同等的权重,这样高表达基因就不会占主导地位,其结果存储在pbmc[["RNA"]]@scale.data中,这里默认ScaleData 进行了中心化,标准化.
## 全局缩放
all.genes <- rownames(pbmc)
pbmc <- ScaleData(object = pbmc, features = all.genes)
head(pbmc[["RNA"]]@scale.data[, 1:3])
## AAACATACAACCAC AAACATTGAGCTAC AAACCGTGCTTCCG
## AL627309.1 -0.06452497 -0.06452497 -0.06452497
## AP006222.2 -0.03726409 -0.03726409 -0.03726409
## RP11-206L10.2 -0.04153370 -0.04153370 -0.04153370
## RP11-206L10.9 -0.03734170 -0.03734170 -0.03734170
## LINC00115 -0.08343360 -0.08343360 -0.08343360
## NOC2L -0.32126699 -0.32126699 -0.32126699
6.2 删除不需要的数据
使用ScaleData函数从单细胞数据集中删除不需要的变量。例如,我们可以“regress out”与细胞周期阶段或线粒体污染相关的异质性(去除不需要的变量,即校正协变量,校正线粒体基因比例的影响)。如下:
# 删除不需要的数据,校正线粒体基因引起的影响
# 这个步骤非常耗时
pbmc <- ScaleData(pbmc, vars.to.regress = "percent.mt")
Tips
我们对矩阵进行了细胞水平质控,但是如何对基因进行质控,当然我们创建seurat 对象时候,已经设定一个cutoff. 基因必须早多少个细胞中表达. 但是如果直接用这些基因进行聚类和降维(T-sne/UMAP 可视化 可能引起偏差,所以需要先进行一下PCA 初步选取主要的主成分,对应着权重不同的基因,再用这些基因进行聚类,降维可视化效果更好)
7. PCA 分析
在PCA分析中,前文标记的高差异基因[email protected]会拿来做分析的数据。当然人们也可以指定自己希望的使用的基因。当细胞数越多,使用标记的高差异基因越接近考虑全基因组时得到的结论。所以,在这一步的时候我们可以看出,之前标记的高差异基因如果数量过低的话,会对这一步的影响很大。
pbmc <- RunPCA(object = pbmc, features = VariableFeatures(pbmc))
## PC_ 1
## Positive: CST3, TYROBP, AIF1, FTL, LST1, FTH1, LYZ, FCER1G, TYMP, FCN1
## S100A9, CFD, LGALS1, CTSS, SERPINA1, S100A8, LGALS2, COTL1, PSAP, IFITM3
## SPI1, SAT1, CFP, S100A11, NPC2, IFI30, LGALS3, GSTP1, S100A6, OAZ1
## Negative: LTB, IL32, IL7R, CD2, B2M, ACAP1, STK17A, CTSW, AQP3, GIMAP5
## RARRES3, CCL5, HOPX, GZMA, SELL, CST7, PPP2R5C, BEX2, CD8B, MYC
## TNFAIP8, ETS1, GZMK, TCF7, ZAP70, RORA, PRF1, KLRG1, GIMAP7, LDLRAP1
......
......
......
## PC_ 5
## Positive: LTB, IL7R, CKB, VIM, AQP3, MS4A7, CYTIP, RGS10, RP11-290F20.3, CD2
## SIGLEC10, HMOX1, HN1, NDUFA4, ATP1A1, TRADD, LILRB2, PTGES3, FYB, ANXA5
## TCF7, CORO1B, FAM110A, IL32, ABRACL, PPA1, TIMP1, GDI2, GIMAP7, VMO1
## Negative: GZMB, FGFBP2, NKG7, GNLY, CST7, PRF1, CCL4, GZMA, SPON2, GZMH
## S100A8, XCL2, CTSW, CLIC3, AKR1C3, CCL5, CCL3, IGFBP7, XCL1, S100A9
## LGALS2, GPR56, HOPX, TTC38, GSTP1, RBP7, TYROBP, CD14, S100A12, MS4A6A
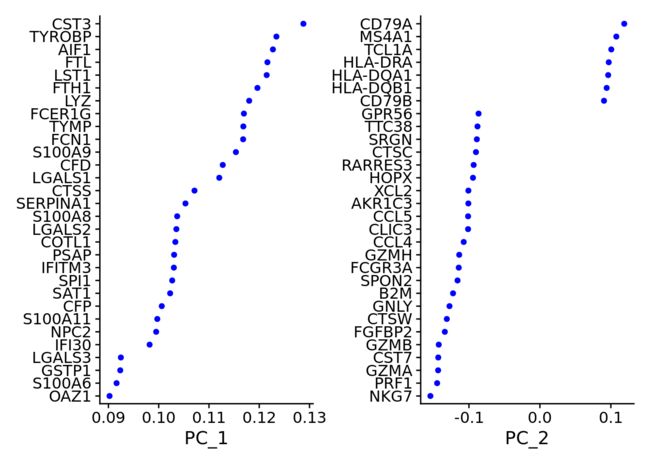
Seurat提供了多种手段来查看PCA分析的结果,其中包括PrintPCA, VizPCA, PCAPlot, 以及PCHeatmap
【一维可视化】 对每一个维度,权重较大的基因进行可视化
VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")
【二维可视化】 将细胞降维到二维空间
DimPlot(object = pbmc, reduction = "pca")
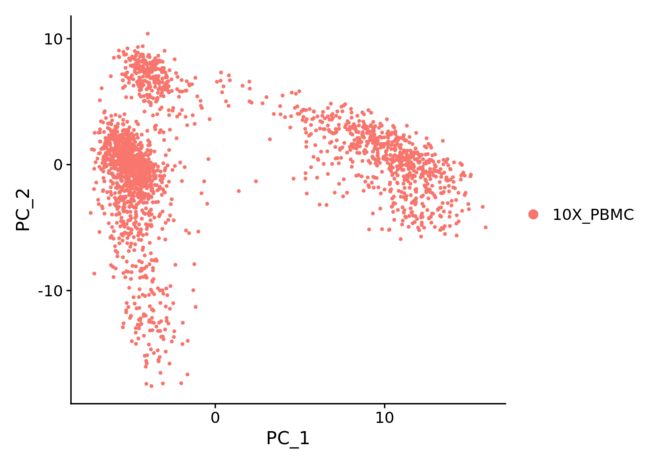
Tips: 下图以PC_1 来看,是解释细胞分布最大的基因组合方向,所有的基因是维度,有些基因对这个方向共享很大,也就是说更容易分开细胞,这些基因可能更加重要。下面是热图1展示这些基因对于区分细胞的效果,可以看到将很多细胞区分成紫色和黄色.效果不错.
可以看到5,6维度貌似区分不开了.
DimHeatmap(pbmc, dims = 1:12, cells = 500, balanced = TRUE)
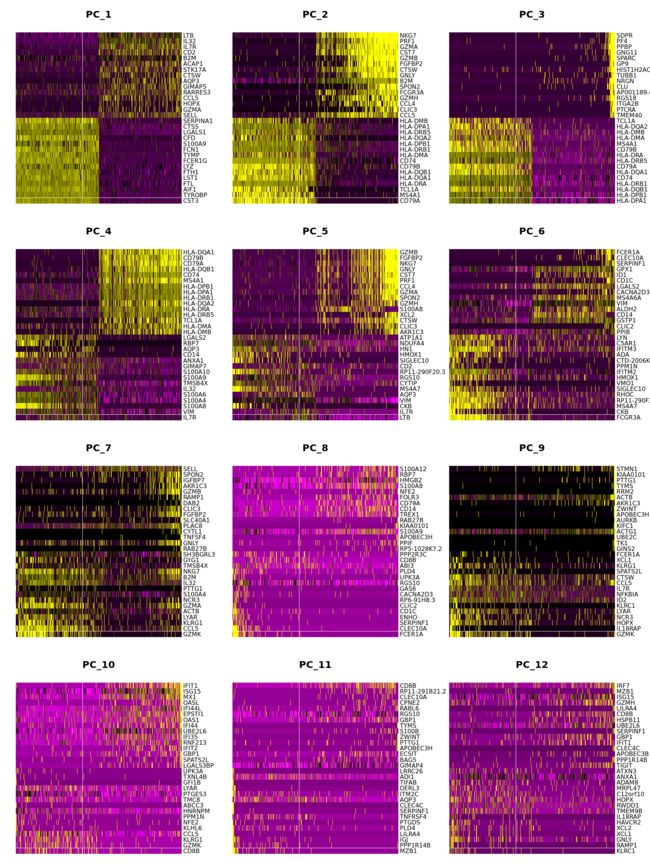
8. 决定需要考虑的PC
Suerat使用了jackStraw方法[4]来估计应该使用多少个principal components的基因来进行下游分析。
# 注意: 这可能是一个非常费时的过程。
pbmc <- JackStraw(pbmc, num.replicate = 100) # 重复一百次
pbmc <- ScoreJackStraw(pbmc, dims = 1:20) # 选择20个PC
JackStrawPlot以及PCElbowPlot函数提供给我们两种可视化手段来查看jackStraw的结果。使用JackStrawPlot可以查看每个PC的p value,这样方便我们通过p值进行筛选。PCElbowPlot可以方便的查看从哪个PC开始,差分度就进入的平台期。
- 1.
JackStrawPlot
JackStrawPlot(pbmc, dims = 1:15)
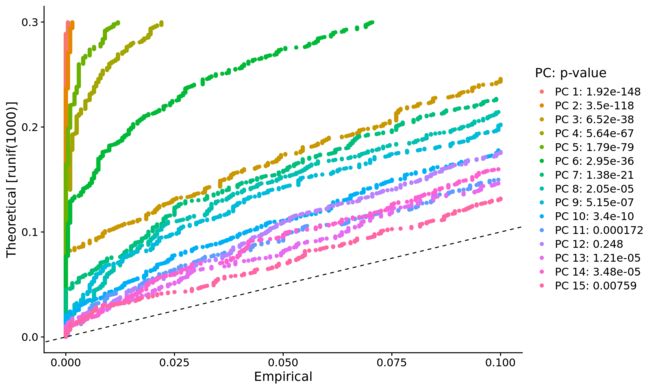
-
2.
PCElbowPlot: 拐点图 # 最大可设置到50ElbowPlot(pbmc,ndims = 50, reduction = "pca") # 最大可设置到50可以直观看到7 后面共享比较平均.
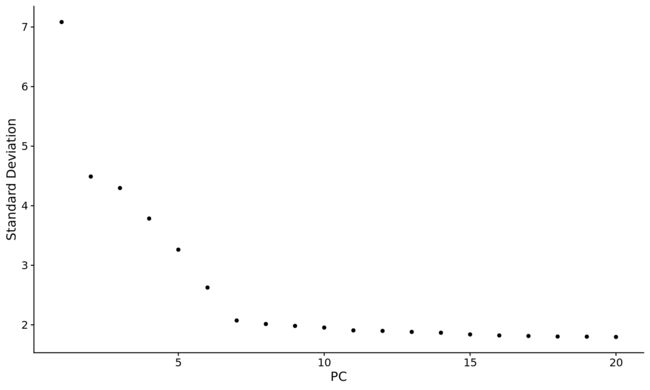
先择PC,对于得到正确的结果具体关键性的作用。除了上面的方法以外,还可以使用其它的一些办法,比如说使用GSEA等富集分析的办法来选择PC,或者说使用一个随机模型来查看具体哪些PC会比较有效果。但是后者这一方法在实现起来需要消耗过多的资源。
在本例中,因为是Seurat挑选的例子,所以通过上面的JackStraw方法,只要把cut.off值设置在7-10之间,就可以得到差不多的结果。
Tips: 确定进行聚类和降维的基因和细胞后,下一步就是聚类了,在这里需要搞情况聚类和降维作用是不一样的。特别是T-sne是降维可视化作用,而不是用它来聚类.为什么看到T-sne 图是不同颜色的色块呢?常见的聚类包括kmeans/hclust .
9.细胞分集
当决定了使用哪些PC中的基因对细胞进行分类之后,就可以使用FindClusters来对细胞分集了。分集后的结果会保存在object@ident slot中。
(Step-1)先在PCA空间中构造一个基于欧氏距离的KNN图,然后根据任意两个细胞在局部邻近区域的共享重叠(Jaccard相似性)来细化它们之间的边权值。此步骤使用FindNeighbors函数执行,并将之前定义的数据集维度作为输入。
(Step-2)使用FindClusters函数对数据进行优化,并包含一个分辨率参数,该参数设置下游集群的“granularity”,增加的值将导致更多的集群。将该参数设置在0.4-1.2之间,对于3K左右的单细胞数据集通常会得到良好的结果。对于较大的数据集,最佳分辨率通常会增加。可以使用Idents函数找到clusters。
下面选取了1到10 维度的基因进行聚类分析
pbmc <- FindNeighbors(pbmc, dims = 1:10) ## dims 作为输入的维度
pbmc <- FindClusters(pbmc, resolution = 0.6) ## 分辨率参数,和cluster 个数有关系,如何你想获得粗糙的分类,需要调成大一些.
# 查看一下前几个的cluster,结果聚成3类.可能发现新的细胞系.
head(Idents(pbmc), 5)
## AAACATACAACCAC AAACATTGAGCTAC AAACCGTGCTTCCG AAACCGTGTATGCG AAACGCTGGTTCTT
## 4 3 0 6 4
## Levels: 0 1 2 3 4 5 6 7 8
10.降维后,用聚类的类别可视化【UMAP/tSNE】
使用UMAP或者tSNE的好处在于将多维的PC图降维至2维图像中来,这样方便分集细胞的可视化。 UMAP皱缩成团分不开,而tSNE图则分散得多。
pbmc <- RunUMAP(pbmc, dims = 1:10)
P1 <- DimPlot(pbmc, reduction = "umap", label = TRUE)
pbmc <- RunTSNE(pbmc, dims = 1:10)
P2 <- DimPlot(pbmc, reduction = "tsne",label = TRUE)
P1 + P2
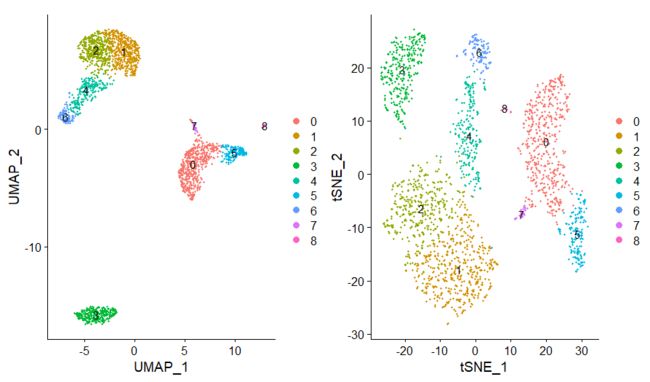
得到聚类分组结果,通常需要继续分析,在每组里面那些标记基因。由于10x genomic 对每一个细胞都进行标签化,不知道每一个标签对应那种细胞,可以通过每组的标记基因对每组进行细胞类型判别(根据文献说明的已知marker),进而发现新的标记基因.
11.寻找分集标记基因
生成了tSNE图像后,我们还需要确认这一堆堆的细胞都分别是些什么细胞,这时,可以使用特定细胞的marker来对不同的细胞群进行标记。但是想要对它们进行标记,首先需要得到每个集群差异表达的基因。
在比较时,Seurat使用一比多的办法。如果需要对cluster 1的细胞(ident.1)进行差异分析时,它比较的对象将是除了cluster 1以外的其它所有的细胞。也可以使用ident.2参数来传递需要比较的对象。 在FindMarkers函数中,min.pct是指的marker基因所占细胞数的最低比例,这里使用1/4。 使用max.cells.per.ident可以让函数对细胞进行抽样,以加速计算。 FindAllMarkers可以一次性找到所有的高表达的marker。
# find all markers of cluster 1
cluster1.markers <- FindMarkers(object = pbmc, ident.1 = 1, min.pct = 0.25)
head(cluster1.markers, n = 5)
## p_val avg_logFC pct.1 pct.2 p_val_adj
## RPS12 4.371171e-118 0.5264969 1 0.990 5.994624e-114
## RPS6 1.681008e-114 0.5234111 1 0.994 2.305334e-110
## RPS27 1.719993e-107 0.5242477 1 0.991 2.358799e-103
## RPS14 1.546910e-105 0.4500125 1 0.993 2.121432e-101
## RPS25 5.848778e-98 0.5427189 1 0.973 8.021015e-94
# find all markers distinguishing cluster 5 from clusters 0 and 3
cluster5.markers <- FindMarkers(object = pbmc, ident.1 = 5,
ident.2 = c(0,3), min.pct = 0.25)
head(cluster5.markers, n = 5)
## p_val avg_logFC pct.1 pct.2 p_val_adj
## FCGR3A 3.849393e-139 2.723391 0.981 0.098 5.279058e-135
## RHOC 1.149445e-91 1.665090 0.865 0.130 1.576350e-87
## CDKN1C 6.799344e-75 1.061123 0.519 0.022 9.324621e-71
## IFITM2 1.347125e-73 1.560680 1.000 0.644 1.847448e-69
## HES4 3.616785e-69 1.107357 0.596 0.052 4.960059e-65
计算所有类和其他类的标记基因
# find markers for every cluster compared to all remaining cells,
# report only the positive ones
pbmc.markers <- FindAllMarkers(object = pbmc, only.pos = TRUE,
min.pct = 0.25, thresh.use = 0.25)
pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_logFC)
## # A tibble: 18 x 7
## # Groups: cluster [9]
## p_val avg_logFC pct.1 pct.2 p_val_adj cluster gene
##
## 1 0. 3.72 0.969 0.136 0. 0 S100A8
## 2 6.30e-298 3.77 0.996 0.236 8.63e-294 0 S100A9
## 3 7.44e- 94 0.807 0.938 0.587 1.02e- 89 1 LDHB
## 4 3.14e- 61 0.848 0.426 0.105 4.31e- 57 1 CCR7
## 5 1.22e- 80 0.975 0.981 0.635 1.67e- 76 2 LTB
## 6 5.02e- 69 0.956 0.783 0.315 6.88e- 65 2 IL7R
## 7 0. 2.95 0.931 0.045 0. 3 CD79A
## 8 5.62e-214 2.46 0.617 0.024 7.71e-210 3 TCL1A
## 9 3.86e-164 2.39 0.973 0.22 5.30e-160 4 CCL5
## 10 2.97e-142 2.12 0.595 0.052 4.07e-138 4 GZMK
## 11 1.66e-171 2.34 0.981 0.135 2.27e-167 5 FCGR3A
## 12 1.68e-106 1.90 1 0.368 2.30e-102 5 LST1
## 13 2.24e-211 3.48 0.972 0.063 3.07e-207 6 GZMB
## 14 1.84e-127 3.52 0.943 0.138 2.52e-123 6 GNLY
## 15 1.11e-180 2.68 0.806 0.013 1.52e-176 7 FCER1A
## 16 5.01e- 20 1.93 1 0.548 6.87e- 16 7 HLA-DPB1
## 17 4.08e-171 5.02 1 0.012 5.60e-167 8 PF4
## 18 6.09e- 99 5.96 1 0.026 8.35e- 95 8 PPBP
还可以使用不同的算法来寻找差异表达的基因。
cluster1.markers <- FindMarkers(object = pbmc, ident.1 = 0,
thresh.use = 0.25, test.use = "roc",
only.pos = TRUE)
想了解更多关于查找差异表达基因的信息,可以查看Seurat的vignette。
【一维可视化】标记基因在不同cluster 表达量
拿到差异表达基因后,可以使用VlnPlot来查看结果。
VlnPlot(object = pbmc, features = c("MS4A1", "CD79A"))
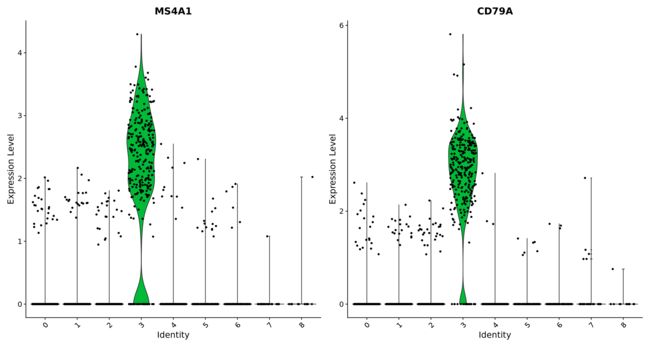
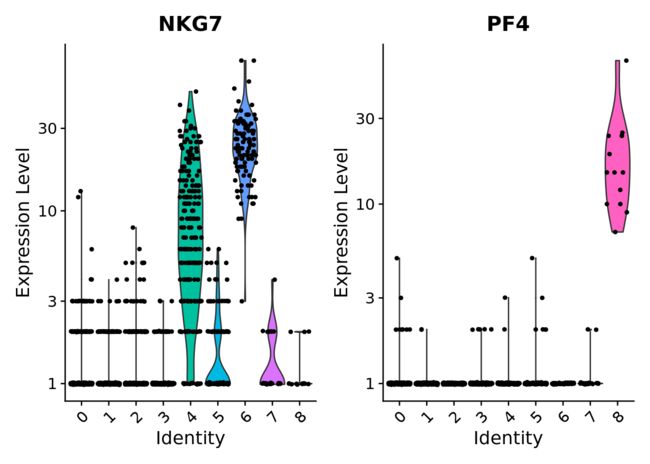
# you can plot raw UMI counts as well
VlnPlot(object = pbmc, features= c("NKG7", "PF4"),
slot = "counts", log = TRUE)
## RidgePlot
RidgePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A"))
## DotPlot
DotPlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A"))
【二维可视化】标记基因在不同细胞表达量
FeaturePlot(object = pbmc,
features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A",
"FCGR3A", "LYZ", "PPBP", "CD8A"),
cols = c("blue", "red"))

提取所有cluster 的top10 标记基因可视化
top10 <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_logFC)
# 设置slim.col.label=TRUE将避免打印每个细胞的名称,而只打印cluster的ID.
DoHeatmap(object = pbmc, features = top10$gene) + NoLegend()

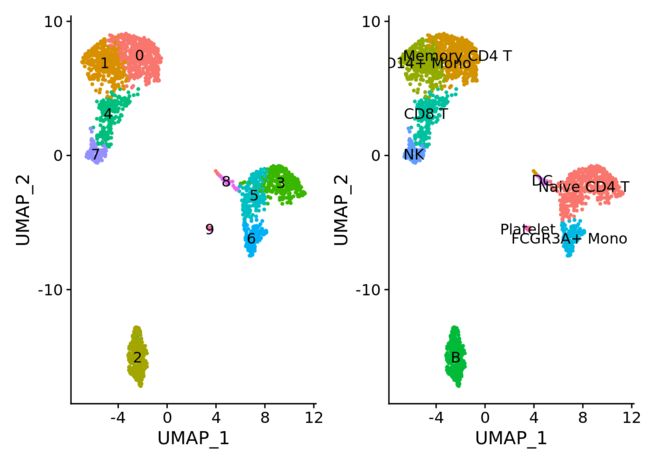
目前我们通过聚类知道了,将细胞分成8个类别,但是并不知道具体是那种细胞。所有需要进一步通过已知的特定细胞标记基因对cluster 打上细胞类型标签.
12.对分集细胞进行标记
对于示例数据,我们已经有一些比较常见的细胞marker了,很方便的就可以对分集的细胞进行标记了。
| Cluster ID | Markers | Cell Type |
|---|---|---|
| 0 | IL7R, CCR7 | Naive CD4+ T |
| 1 | IL7R, S100A4 | Memory CD4+ |
| 2 | CD14, LYZ | CD14+ Mono |
| 3 | MS4A1 | B |
| 4 | CD8A | CD8+ T |
| 5 | FCGR3A, MS4A7 | FCGR3A+ Mono |
| 6 | GNLY, NKG7 | NK |
| 7 | FCER1A, CST3 | DC |
| 8 | PPBP | Platelet |
将原来的cluster 1-8 ,修改成对应的细胞名称
new.cluster.ids <- c("Naive CD4 T", "Memory CD4 T",
"CD14+ Mono", "B", "CD8 T",
"FCGR3A+ Mono",
"NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
P1 <- DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
P2 <- DimPlot(pbmc, reduction = "tsne", label = TRUE, pt.size = 0.5) + NoLegend()
P1 + P2

当我们修改了聚类的参数,结果会不同。比如分辨率参数.
# 生成一个快照, 其实就是把pbmc@ident中的数据拷贝到[email protected]中。
# 当我们想把[email protected]中的值赋值给pbmc@ident的话,可以使用SetAllIdent函数。
# 比如pbmc <- SetAllIdent(object=pbmc, id = 'orig.ident')
pbmc <- StashIdent(object = pbmc, save.name = "ClusterNames_0.6")
# 现在我们使用不同的resolution来重新分群。当我们提高resolution的时候,
# 我们可以得到更多的群。
# 之前我们在FindClusters中设置了save.snn=TRUE,所以可以不用再计算SNN了。
# 下面就直接提高一下区分度,设置resolution = 0.8
pbmc <- FindClusters(pbmc, resolution = 0.8)
分辨率左图是0.8,右图0.6
# 并排画两个tSNE,右边的是用ClusterNames_0.6 (resolution=0.6) 来分组颜色的。
# 我们可以看到当resolution提高之后,CD4 T细胞被分成了两个亚群。
plot1 <- DimPlot(object = pbmc, reduction = "umap", label = TRUE) + NoLegend()
plot2 <- DimPlot(object = pbmc, reduction = "umap", label = TRUE,
group.by = "ClusterNames_0.6") +
NoLegend()
plot1 + plot2
类别不同,我们又可以按照这一次聚类结果,寻找marker基因
# 我们再一次寻找不同分集中的marders。
tcell.markers <- FindMarkers(object = pbmc, ident.1 = 0, ident.2 = 1)
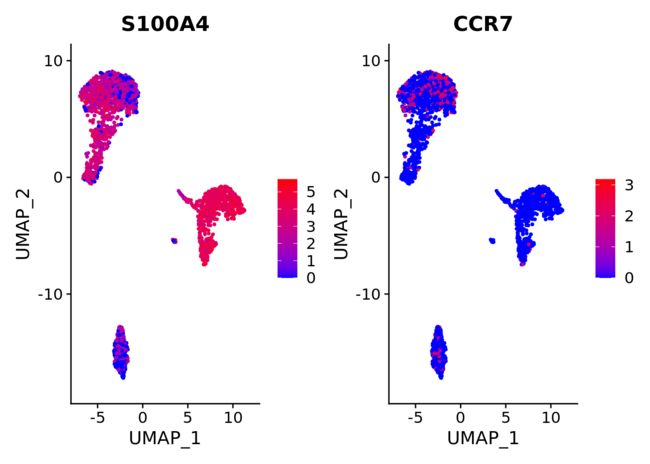
# Most of the markers tend to be expressed in C1 (i.e. S100A4).
# However, we can see that CCR7 is upregulated in
# C0, strongly indicating that we can differentiate memory from naive CD4 cells.
# cols.use demarcates the color palette from low to high expression
FeaturePlot(object = pbmc, features = c("S100A4", "CCR7"),
cols = c("blue", "red"))