转发自http://crickcollege.com/news/155.html
缘起
在2016年三月的Molecular & Cellular Proteomics 上有篇名为"Large Scale Mass Spectrometry-based Identifications of Enzyme-mediated Protein Methylation Are Subject to High False Discovery Rates" (http://dx.doi.org/10.1074/mcp.M115.055384)的文章来自于新南威尔士大学,采用了三种样品制备方式(coomassie gel, unstained gel, HILIC)与三种离子化方式(CID, ETD, HCD)组合,得到了9组数据,对甲基化肽段进行了非常翔实的研究。文章指出了数据分析中一个比较严重的问题:可怕的FDR值!甲基化肽段的FDR有70%到90%,而非修饰肽段的target/decoy比例仅仅为1%!太可怕了吧!

对FDR含义还有疑惑的同学请参考往期推文:
p值、E值、FDR、q值…你晕菜了吗?
人气推文p值、E值、FDR、q值…你晕菜了吗?续集来啦!
为何此类肽段的FDR如此之高?!是不是意味着修饰组学中应用target/decoy策略会存在一些原理上的缺陷呢?
对于这个问题需要分好几个方面来讨论。首先,我们需要确认从整个结果中提取一部分肽段来进行分析是否恰好也能用全局的FDR来反映其假阳性率;然后我们需要探讨数据库搜索中位点置信度分析的缺陷,以及混合多种搜库结果时target/decoy统计相关的一些问题。
全局FDR?
这篇MCP文章重点关注的是甲基化肽段:是否发生了后修饰以及位点信息是否正确。作者用了多种可变修饰组合和结果的合并方式来进行搜库。所有搜库方式均将Carbamidomethyl (C) 和Oxidation (M) 设为可变修饰,然后与下列组合进行一同搜库:
1. Methyl (K), Dimethyl (K), Trimethyl (K)
2. Methyl (R), Dimethyl (R)
3. Methyl (DE)
4. Ethyl (DE)
5. Propyl (DE)
6. Propionamide (C)
那么问题来了,如果将全局的PSM FDR设为1%,我们能否假设甲基化肽段的FDR也是1%?
其中一种方法就是将甲基化肽段的Target数据和decoy数据进行比较。在作者上传的数据共享集 PRIDE PXD002857(http://www.ebi.ac.uk/pride/archive/projects/PXD002857)中并没有包含原始的搜索结果,于是我们取了其中一个数据集 (nostainbands_orbi_1.raw through 28),采用作者提供的参数和修饰设置中的第一组在Mascot软件中进行了测试。由于软件、参数、数据库版本无法完全相同,搜库结果和原文不完全一致,但十分近似。我们采用Mascot expect打分作为target/decoy分析对象并卡了PSM 1%FDR。下表列出了相关打分结果Target和decoy的一些统计信息。其中的615个decoy结果我们假定是错误的,而61,531 target结果我们假定其中99%是正确的。
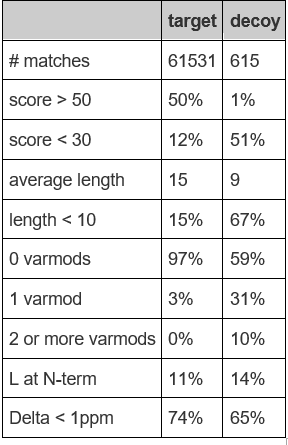
通常我们说的1% FDR是针对整个数据集的结果,而不是针对单个匹配的。相对于高得分来说,低得分匹配很可能是错误的。如果我们从低于30分的结果中取出一部分结果看它们的FDR,那么其FDR一定高于1%,而从50分以上结果中取出一部分的话,一定是小于1%FDR的。从上面表格可以看得非常明显。比如,99%的错误匹配都低于50分。
如果我们提取数据子集的标准和得分有一定的相关性,那么结果一定也是类似的。表格中我们可以看到较短的肽段的FDR会明显过高。平均正确肽段的长度是15,而错误匹配的是9,而且67%的错误匹配都短于10个氨基酸,正确的肽段中相应比例才15%。这是因为肽段打分与其长度相关,越长的肽段能够得到越多的离子匹配。因此,数据子集如果选择的是9个氨基酸以下的肽段,会得到极高的FDR值。
同样的,可变修饰的数量也是非常重要的因素。正确结果中只有3%的匹配包含可变修饰,而41%的错误匹配包含可变修饰。这并不是因为可变修饰的数量和肽段得分有关,而是因为随机匹配中可变修饰的可能性太多了。
例如,SwissProt数据库中K(Lysine,赖氨酸)和R(Arginine,精氨酸)的比例是相近的,所以我们可以预估酶切肽段中KR比例也相近。但3种可变修饰都修饰在K上,因此任何一个包含K的非修饰肽,都有3种备选修饰去参与打分。同时,将漏切数量设为2,也就是说每个肽都可能包含1个或2个K/R。那么对于一个包含2个K的肽段来说就有15种可变修饰组合了,而3个K的肽段来说有63种组合。
以上的组合爆炸并不是甲基化独有的,对于所有可变修饰来说都或多或少有些影响。因此,我们洞察到:不!修饰肽的FDR不可能和全局FDR一样。

我们再回头看下表格的最后两行,这两个参数下的target和decoy匹配基本一样多。亮氨酸在肽段末端与否和肽段打分完全扯不上关系,因此提取子集的时候以此为参数是十分的安全的,虽然并没什么意义。另一个结果反而会让很多人感到吃惊。我们会觉得在超高精度的时候正确匹配的比例应该大于错误匹配,但现在看来并非如此。
修饰态的FDR
回到正题,有啥好办法来解决修饰肽的FDR计算问题呢?如果这是我们的实验主要目的,我们可能得牺牲一些灵敏度来提高显著性阈值。比如这组数据如果我们大大提高significance p的阈值到0.0008,则修饰肽的FDR降到了1%,同时全局的FDR就只有0.12%了。
在这篇MCP文章里,作者说单纯提高打分阈值没法得到一个合适的FDR值,他采用了一系列更严苛的标准来过滤结果。再进一步来说,在得到正确匹配的同时还要对其修饰位点进行评估则是更加困难的工作了。我们好不容易对肽段匹配进行了FDR过滤,发现还得对位点进行过滤。否则严格来说,这还是个错误匹配。这个时候如何来定义错误匹配又是个问题。我们就此讨论如下几个问题。
作者采用了非常规的正确和错误匹配定义
本文主要研究对象是甲基化肽段,目标之一是区分修饰肽到底修饰在了正确的残基上还是可能为一个假的匹配。
作者在酵母细胞培养的过程中引入标记的甲硫氨酸来协助判断正确的甲基化位点。比如其中一个来自于Elongation factor 1 – alpha的正确匹配肽段NVSVK*EIR,其赖氨酸可能发生单、双、三甲基化。让我们假定搜库时采用本文开头提到的第一套搜库修饰组合,结果发现在K5上得到一个高得分匹配的二甲基化修饰。那么可以确信的是,当作者采用第4套修饰参数时也会得到一个E6上修饰的较低得分但依然可信的乙基化匹配。在原文中,作者认为发生此类矛盾结果的匹配会是假阳性匹配。也就是说,如果在第二套修饰组合匹配时得到一个R8的二甲基化结果,那么这也会被认为是假阳性。
这个策略虽然看上去可以避免一些假阳性结果,但并不客观。实际样品中乙基化DE的比例越高,则K/R上匹配二甲基化的错误率也就越高。实际情况中,两者并无关联。但作者为了避免假阳性,还做了类似的一些假定,比如靠近如果靠近半胱氨酸的位置发生甲基化,那么很有可能和假的Propionamide (C)错配有关,因为他们的组合等于Carbamidomethyl (C)。
修饰会带来结果的不确定性
让我们回顾下正确和错误匹配的定义。比如Mascot,就以一个匹配是否是随机事件来评估结果正确的可能性。换句话讲,也就是某个谱图匹配是否可能得到一个完全不相干的序列结果。
当两条序列非常相似的时候,会发生什么呢?比如一条很长的肽段得到了一个高得分、高可信度的匹配结果。如果我们随机交换其中两个相邻氨基酸的顺序,谱图匹配得分很难说会改变多少。很多情况下它依然会得到一个高得分。幸运的是我们不会在常规搜库中经常遇到这样的突变。
假阳性匹配更多的发生在一个SNP加一个修饰和发生分子量相当的两个修饰这样的两种结果的比较中。这种情况下母离子质核比是不变的。比如,分析对象包含这样的序列: —-MA—- 而数据库里的序列则是—-MS—- ,其中M是指甲硫氨酸氧化。严格的讲这是一个错误匹配,但实际打分软件无法区分这样的差异,会对这两个结果给出相同的打分。这两条序列并非完全无关,如果两者均出现在一个数据库里,你只能祈祷正确匹配的结果能比另一个得分高那么点,虽然更多的可能是完全相等。
以上情况同样会发生在修饰检测中。检测一条肽段是否发生修饰是很简单的,因为会有非常准确的质量差异。但产生一个特定分子量的质量偏移的原因实在太多了,有时候就算选错了修饰,我们也可能得到一个非常好的匹配结果。比如搜索Phospho (ST)却得到一个高可信的磺酸化匹配,或者搜索methyl (K)却发现隔壁氨基酸的D或E上的修饰结果打分更高。
就算我们只搜索认为正确的修饰组合,接下来要确定修饰位点的难度可能比找到修饰肽更困难。一条肽段很可能是有连续的S或T残基而其在谱图中的差异仅仅会体现在单根谱峰上,而且往往信号很弱。当然,你可以用Mascot 得分进行位点分析,但修饰位点越靠近,置信度分析越难。
Target/decoy方法可以告诉我们的和无法告诉我们的
当我们用target/decoy方法来评估FDR,假阳性结果的计数受限于显著结果中无关序列的比例。由于decoy数据库不包含正确序列或者与正确序列高度同源的序列,FDR评估无法代表以下类型的假阳性:
1. 高度同源的序列同时在搜库容差范围内在加上或去掉一个错误修饰后分子量完全和正确序列一致
2. 序列正确、修饰正确,但位点判断错误,出现在隔壁的残基位点
3. 正确的序列但鉴定到错误的修饰或修饰组合,但该修饰与正确修饰间的质量差异又在容差范围内,比如磷酸化 vs 磺酸化或者 propionamide vs. carbamidomethyl + methyl
4. 正确的序列和修饰的元素组成,但修饰结构错误,如dimethyl vs. ethyl
5. 正确的序列带有错误来源的修饰,比如post-translational vs. artefactual
如果你运气够好得到了正确的匹配,但错误匹配的得分,如以上1-3例子中的结果,很有可能得分非常接近正确匹配,且同时都超过了显著性阈值。当然后续结果报告时你还是幸运的得到了排名第一的正确匹配。但数据库搜索无法区分4-5两种情况的假阳性。
竞争者是必须的
如果MCP的文献作者在设计结果过滤策略时允许出现结果匹配的矛盾性,同时最终只取结果排名第一的结果,那么相应的FDR则应该会降低。在合并结果时,我们可以模拟出一些竞争性的匹配结果,但更简单的方式是用Mascot的容差搜索选项来寻找任何潜在的未知修饰。容差搜索会自动搜索Unimod数据库中的所有可能修饰类型,不过只能匹配肽段上出现一种未知可变修饰的情况。当然出现两种以上未知修饰的比例是非常低的,比如原文献的甲基化匹配的Suppl数据中,我们只发现了一条这样的肽段。 (Tables SII and SIII): NDYGPPRGSYGGSR*GGYDGPR (R7上的methyl和R14上的dimethyl)。
我们用原文的原始数据nostainbands_orbi_1.raw到28.raw测试了Mascot容差搜索是否能够应用于此类研究,设定了固定修饰Carbamidomethyl (C) 和可变修饰Oxidation (M) 及Propionamide (C) 。初次搜库时,FDR阈值设为1%。
我们用实际数据举个例子来说明为何要保留匹配结果的竞争者。比如原文献中有的一个显著匹配结果AEQLYEGPADDANCIAIK:(
在nostaingels_orbi_metK_yeast_psms.txt:
C14找到了22 x Carbamidomethyl 谱图
K18上找到了6 x Methyl, C14找到一个Carbamidomethyl
在nostaingels_orbi_metR_yeast_psms.txt:
C14找到了22 x Carbamidomethyl
在nostaingels_orbi_metDE_yeast_psms.txt:
C14上找到22 x Carbamidomethyl
D11上找到7 x Methyl,C14上找到1个 Carbamidomethyl
匹配到Methyl (K)的结果被认为是假阳性。在容差搜库结果中,由于C14上Propionamide匹配的得分总是比Methyl匹配的得分高,没有一张谱图的结果 Methyl (K)位于打分排名第一,容差搜索结果如下:
22 x Carbamidomethyl on C14
1 x Cys->Dha on C14
4 x Dehydrated on D11, Carbamidomethyl on C14
3 x Deamidation on N13, Carbamidomethyl on C14
6 x Propionamide on C14
1 x Oxidation on Y5, Carbamidomethyl on C14
4 x Carbamidomethyl on D10, Carbamidomethyl on C14
原文献最终采纳了K和R上的甲基化修饰。而容差搜索中找到了39个肽段C端甲基化,而数据库搜索结果并无法区分C端甲基化还是在R侧链。对这些匹配结果,我们参考原文设定的规则,单甲基化不影响酶切而di- 或 tri-甲基化会导致漏切,因此后者修饰一定会发生在C端酯基。这样的话就剩下了91个可能正确的匹配(如Tables SII和SIII) 而另外24个K或R的甲基化匹配被认为是错误的。
这样的策略比完全去掉竞争匹配会好些,但依然离1%的FDR非常遥远。原文中,甲基化FDR只计算不同的肽段而不是PSM,这种方法又引出了另一个复杂的问题:FDR计算基于 counts of matches (PSMs) 还是distinct sequences
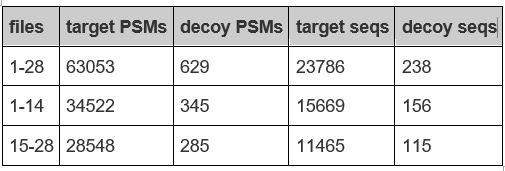
上表格统计了28个文件总体FDR分布,以及前14个和后14个分别统计的结果。搜索参数完全相同。当我们以PSM FDR为标准统计时,前一半数据和后一半数据是可以直接加和的。而如果以Peptide FDR为标准计算时则无法简单相加。原因很简单:一部分序列是两个数据集共有的。如果你想合并多组数据的搜库结果又同时使用的是Peptide FDR算法,1%FDR阈值需要重新计算,然而如果你在两组数据中使用的参数不完全相同,这种计算又失去了科学性。
错误序列的数量受数据库规模的限制,当然通常其数量大于正确匹配的序列。假定,我们想重分析某一个数据,同时不停地加入其技术重复的结果,直到我们发现正确匹配序列的数量不再增加。这时候是否意味着再加入额外的数据结果反而因为新加入的候选匹配都是假阳性的从而会导致FDR升高么?不,因为谱图打分或者expect值只会显示最佳匹配的结果。当加入更多数据时,正确匹配的score (或expect值) 分布会逐渐往高得分方向偏移,其速度一定快于错误匹配的得分分布偏移。你可以将其看成是结果谱图的信噪比得到了逐渐提高。每张新谱图的加入同时提高谱峰信号和基线信号,但由于谱峰信号的强度一定高于基线,因此不停累加谱图的话,结果的信噪比一定是越来越好的。
一般来说基于Distinct Peptide FDR 1%的算法往往比PSM FDR更严格。也就是说如果你设定PSM FDR 1%,相应的Peptide FDR不会也是1%。(当我们提到distinct sequence时,既可以认为只考虑其序列的独特性,也可以进一步将其修饰状态、电荷数或其他信息都一并纳入计算。)
在原文中,全局FDR是基于Percolator q-value < 0.01来计算1%的PSM FDR。而对于甲基化肽段,看上去结果又是以Mascot expect 值< 0.05来过滤的。某些角度来说这两种方法共用会导致结果解释不清楚,但可以确定的是这套标准的确是基于PSM计算而不是Distinct Peptides来的。换句话说,甲基化肽的FDR是基于对‘non redundant PSMs’计数来计算的,也就是基于distinct sequence +修饰状态的组合。原文基于PSM设定阈值而报告FDR的时候却是基于distinct sequences的方式是有问题的。对我们来说最好在同一个研究中使用其中一种策略不变,尤其是对于正确匹配肽数量不多而谱图非常多的研究。比如原文只有共59个正确匹配(Tables SII 和 SIII)。
总结
总的来说,该文章是一片非常重要而研究透彻的文献,其反应了数据库搜索策略中的一些缺陷:
• 全局FDR和提取子集,如修饰肽的FDR一定不一样
• Target/decoy 评估了结果匹配是无关序列的可能性。但无法将其应用于区分高度同源肽段或者区分修饰组合匹配的情况。
• 我们需要依赖于多个候选匹配间得分的互相竞争来排除一些假阳性
• 数据库搜索不能区分元素组成正确而结构不对的修饰
• FDR计算可以基于PSM也可以基于distinct sequence但一定不要将其在同一篇文章中混用
• 如果你采用基于distinct sequences FDR的策略,那么最好不要尝试合并多个的搜库结果。
最后,我们也非常同意作者的观点,高度修饰肽的组学数据光是采用数据库搜索和FDR评估的方式来进行结果可信度评估,目前来说还远远不够成熟。