写在前面:
这部分主要做一些数据可视化,富集分析暂时放下一部分,如果想跳过这里,请直接移步RNA-seq(9):富集分析
---------------------------------------------------
参考资料:
Analyzing RNA-seq data with DESeq2
[Count-Based Differential Expression Analysis of RNA-seq Data]
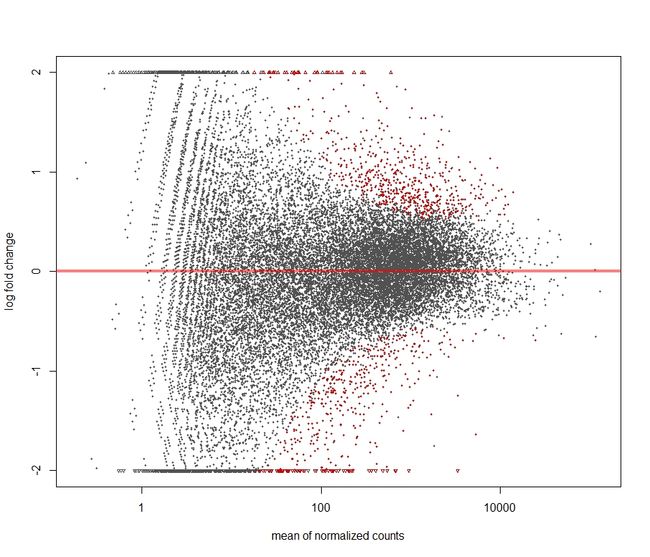
1 MA plot
An MA plot is an application of a Bland–Altman plot for visual representation of genomic data. The plot visualizes the differences between measurements taken in two samples, by transforming the data onto M (log ratio) and A (mean average) scales, then plotting these values. Though originally applied in the context of two channel DNA microarray gene expression data, MA plots are also used to visualise high-throughput sequencing analysis.
MA这部分代码主要参考hoptop,并进行修改
In DESeq2, the function plotMA shows the log2 fold changes attributable to a given variable over the mean of normalized counts for all the samples in the DESeqDataSet. Points will be colored red if the adjusted p value is less than 0.1. Points which fall out of the window are plotted as open triangles pointing either up or down.
- 没有经过 statistical moderation平缓log2 fold changes的情况
plotMA(res,ylim=c(-2,2))
topGene <- rownames(res)[which.min(res$padj)]
with(res[topGene, ], {
points(baseMean, log2FoldChange, col="dodgerblue", cex=6, lwd=2)
text(baseMean, log2FoldChange, topGene, pos=2, col="dodgerblue")
})
结果如下:
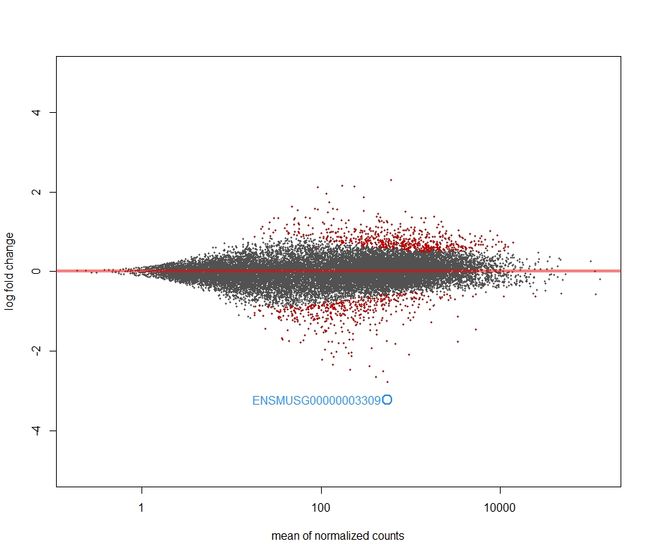
- 经过lfcShrink 收缩log2 fold change
It is more useful visualize the MA-plot for the shrunken log2 fold changes, which remove the noise associated with log2 fold changes from low count genes without requiring arbitrary filtering thresholds.
注意:前面res结果已经按padj排序了,所以这次要按照行名升序再排列回来,否则和dds不一致
res_order<-res[order(row.names(res)),]
res = res_order
res.shrink <- lfcShrink(dds, contrast = c("condition","treat","control"), res=res)
plotMA(res.shrink, ylim = c(-5,5))
topGene <- rownames(res)[which.min(res$padj)]
with(res[topGene, ], {
points(baseMean, log2FoldChange, col="dodgerblue", cex=2, lwd=2)
text(baseMean, log2FoldChange, topGene, pos=2, col="dodgerblue")
})
2 Plot counts
DESeq2提供了一个plotCounts()函数来查看某一个感兴趣的gene在组间的差别。counts会根据groups分组。更多的参数请输入命令?plotCounts下面我们来看plot两个genes
- 一个是padj最小的gene
- 一个是
直接用plotCounts命令
# 不画图,只显示数据
plotCounts(dds, gene=which.min(res$padj), intgroup="condition", returnData=TRUE)
#只画图,不显示数据
plotCounts(dds, gene="ENSMUSG00000024045", intgroup="condition", returnData=FAULSE)
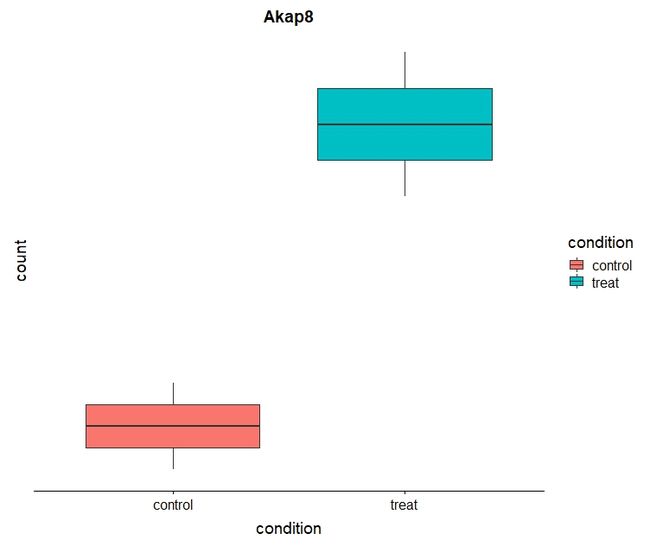
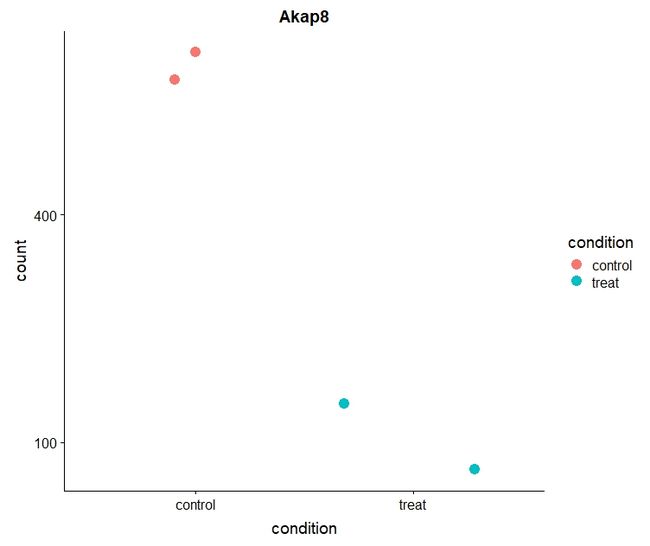
下面用ggplot来画Akap8的box图和point图
- boxplot
# Plot it
plotCounts(dds, gene="ENSMUSG00000024045", intgroup="condition", returnData=TRUE) %>%
ggplot(aes(condition, count)) + geom_boxplot(aes(fill=condition)) + scale_y_log10() + ggtitle("Akap8")
- point plot
d <- plotCounts(dds, gene="ENSMUSG00000024045", intgroup="condition", returnData=TRUE)
ggplot(d, aes(x=condition, y=count)) +
geom_point(aes(color= condition),size= 4, position=position_jitter(w=0.5,h=0)) +
scale_y_log10(breaks=c(25,100,400))+ ggtitle("Akap8")
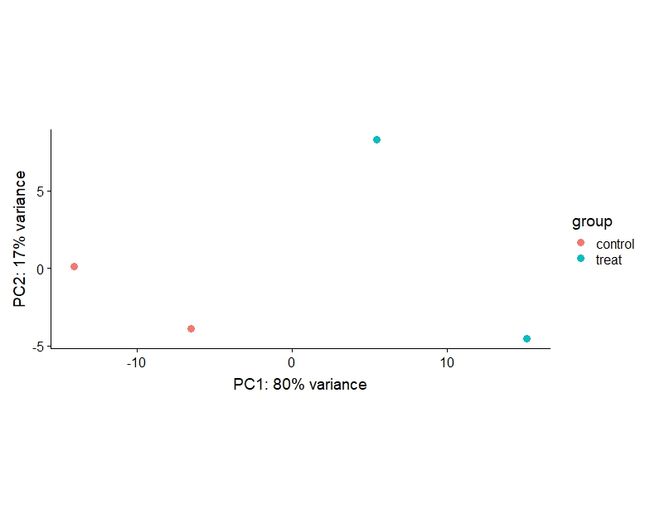
3 PCA(principal components analysis)
- 上面的分析,我们使用的原始的counts数据。但是又一些下游其他分析比如热图(heatmap), PCA或聚类(clustering)我们需要data的转换后的格式,因为如何最好的计算未转换的counts的距离测度仍然不清楚。一个选择是进行log变换。但是因为很多samples的count为0(这意味着 log(0)=−∞,当然也可以使用家counts,比如y=log(n+1)或更普遍使用的y=log(n+n0 ),n代表count值,n0是某个正常数。
但是也有一些其他的方法提供更好的理论矫正,其中有一个称为variance stabilizing transformation(VST),它消除了方差对mean均值的依赖,尤其是低均值时的高log counts的变异。 - DESeq2提供了plotPCA函数进行PCA分析。
?plotPCA查看帮助文件。
用法如下
vsdata <- vst(dds, blind=FALSE)
plotPCA(vsdata, intgroup="condition")
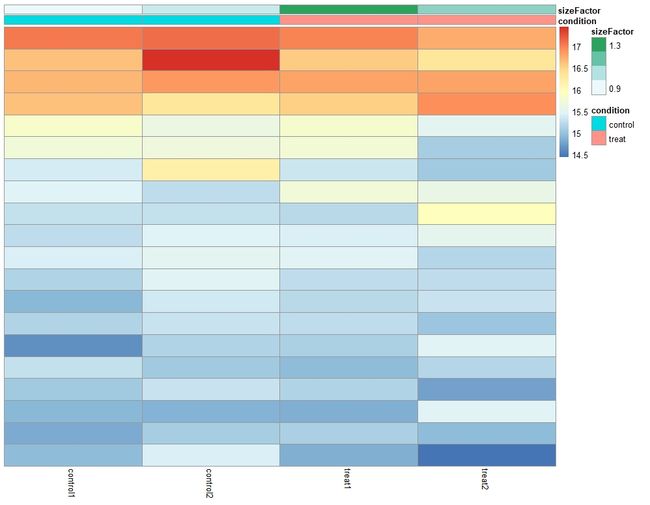
4热图:两部分
4.1 count matrix 热图
根据不同的数据转换方式,可以产生不同类型的heatmap
library("pheatmap")
select<-order(rowMeans(counts(dds, normalized = TRUE)),
decreasing = TRUE)[1:20]
df <- as.data.frame(colData(dds)[,c("condition","sizeFactor")])
# this gives log2(n + 1)
ntd <- normTransform(dds)
pheatmap(assay(ntd)[select,], cluster_rows=FALSE, show_rownames=FALSE,
cluster_cols=FALSE, annotation_col=df)
上面这两幅图看起来没什么区别,我暂且只放一张
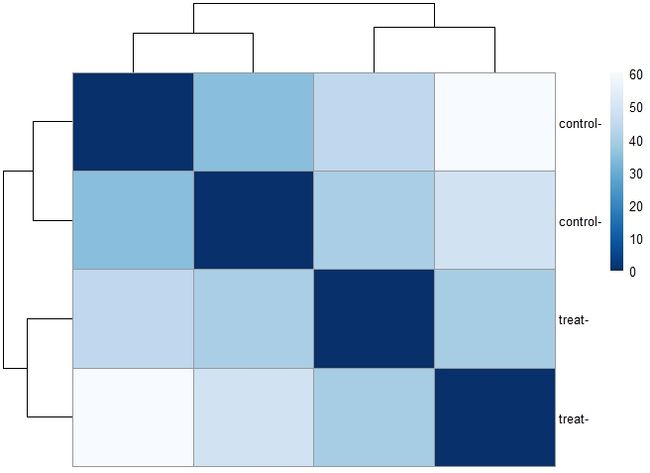
4.2 sample-to-sample distances热图
- 转换数据还可以做出样本聚类热图。用
dist函数来获得sample-to-sample距离。距离矩阵热图中可以清楚看到samples之间的相似与否的总概。需要给heatmap函数基于sample距离提供等级聚类hc。
#sample to sample heatmap
sampleDists <- dist(t(assay(vsd)))
library("RColorBrewer")
sampleDistMatrix <- as.matrix(sampleDists)
rownames(sampleDistMatrix) <- paste(vsd$condition, vsd$type, sep="-")
colnames(sampleDistMatrix) <- NULL
colors <- colorRampPalette( rev(brewer.pal(9, "Blues")) )(255)
pheatmap(sampleDistMatrix,
clustering_distance_rows=sampleDists,
clustering_distance_cols=sampleDists,
col=colors)