我们都知道,从数据质控开始已经进入了scRNA分析阶段,在这个阶段开始测试代码,进行实操是很重要的。测试过程中出现的各种问题可能成为你学习路上的拦路虎。作图丫为大家总结单细胞数据分析时,标准化处理的方法和策略。
序言
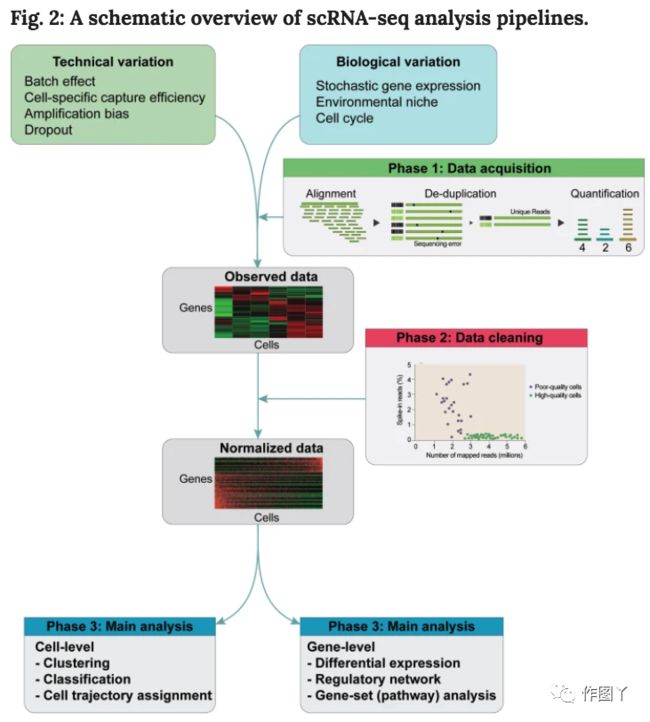
上一期我们介绍了如何对scRNA-seq数据进行预处理与质控,在得到高质量的barcode by cell计数矩阵之后,我们需要通过基因在不同细胞间的差异表达来对细胞聚类。而数据的标准化(normalization)对于准确的比较细胞间的基因表达则是非常重要且必要的一步。今天我们就来一起了解一下如何对scRNA-seq数据标准化。
计数矩阵标准化的必要性
在scRNA-seq中,由于每个细胞的起始转录分子量有限,每个细胞中转录本的捕获以及扩增效率都会有技术差异,因此很难保证样本之间在文库制备上保持高度的一致性。这也造成了多个样本的测序数据中会存在由于文库测序覆盖率(sequencing coverage) 不同而引入的系统差异。数据的标准化目的就是消除这些差异,使得我们得到的分析结果不受技术噪音的影响。
数据标准化一般分为两大类:
· 样本内的标准化(within-sample normalization): 针对由基因特异性(如基因长度,GC content)产生的偏差,标准化后使得同个样本内的基因表达具有可比性。我们常用的RPKM,FPKM 和TPM就是标准化之后的表达值。
· 样本间的标准化(between-sample normalization): 针对样本间的差异,例如测序深度和转录本捕获率,标准化后的表达值能被用于不同样本间的比较。
在scRNA-seq分析中,我们将每个细胞视为一个独立的样本,来比较细胞内不同基因的表达。我们前期的文章提到过,在droplet-based的方法中,只对转录分子的5' 或 3'端测序,因此此类数据的标准化不需要考虑基因长度的影响,对应的就是方法类别就是样本间的标准化。
目前已经有很多针对bulk RNA-seq 数据的成熟的标准化方法,有些方也被用于单细胞分析。然而需要注意的是,由于scRNA-seq数据的高度稀疏性(sparsity) 和技术噪音,直接使用bulk RNA-seq的方法使得对单细胞数据中的低表达基因造成过度矫正。
下面我们主要给大家介绍两大类用于scRNA-seq的标准化方法[2,3]。
第一类常用标准化方法-log-normalization
大家比较熟悉的标准化方法是scaling。由于每个细胞的总计数(也可称为测序深度)不同,首先通过总计数对每个细胞估算出一个size factor,它代表了细胞间由不同测序深度带来的相对偏差值, 然后对每个细胞的总计数除以特定的size factor,以此来达到消除偏差的目的,得到“normalized expression values”用于下游分析。如果在scRNA-seq中用到了spike-ins 或 UMIs,标准化的操作则要根据它们的结果来进行调整。一些用于scRNA-seq的方法有:
· CPM (counts per million) normalization: 这个方法假设所有细胞包含等量的mRNA分子,测序深度的差异仅来源于抽样,即相对偏差全部都体现在细胞的不同计数总和上;因此估计的size factor与细胞计数总和成正比。这个方法在bulk RNA -seq中也很常用。方法对应的R包: Seurat [3],scater [4] 。
· High-count filtering CPM: 是在CPM的基础上,考虑到少数高表达基因对细胞偏差估计的影响,在估算size factor时剔除细胞中表达量高于5%的总计数的基因。
· Scran: 针对单细胞测序的dropout和0计数现象,scran通过合并 (pool) 总计数类似的细胞,通过它们的计数总和来估算一个size factor,然后将其进一步分解,用到每个细胞表达谱的标准化中。R包: scran [5]。
· BASiCS: 基于spike-ins来推断细胞特异的size factor。R包: scRNAseq [6]。
以上这些方法都基于一个假设:对于样本中所有细胞,它们的转录分子量都是相同的。这样同一个size factor才能被用于细胞中的所有基因。
在标准化之后,计数矩阵还需要做log(x+1) 转化。由于在衡量表达值差异大小的时候,我们通常使用的是表达值的对数倍变化(log-fold change),因此需要对计数矩阵作进一步的对数转化。并且由于很多下游的分析工具 (例如差异表达分析) 都假设数据是正态分布的,然而我们知道scRNA-seq数据实际上并不一定满足,因此对数转换则能帮助我们降低数据的skewness,尽管方法比较粗糙但是对之后的分析很实用。
在log(x+1) 转化中的+1是加上的一个伪计数(pseudo-count) ,用来避免未定义的数值0。伪计数的选择比较多,用+1的原因是能保留原始矩阵中的sparsity,即原始表达值为0的在对数转换后仍然为0。当然,你也可以选择其他的数值,如果选择较大的伪计数,低表达基因之间的对数倍变化则会变小,使得下游的差异分析结果由高表达基因主导;反之选择较小的伪计数则能增加低表达基因在差异分析结果中权重。大家可以根据自己的研究目的来调整选择的参数。
以上两步(scaling & log-trans) 结合起来通常被称作“log-normalization” ,这类方法比较简单并且常用。
第二类常用标准化方法- probabilistic model based approach
另外一类标准化方法比较新颖,也更加复杂,是通过拟合分布来对细胞计数构建模型(model molecule counts using probabilistic approaches),用模型拟合的残差 (residuals) 作为基因表达的标准化定量。一些新的基于UMIs的方法,它们的建模主要是使用NB distribution以及zero-inflation NB distribution (ZINB)。一些常见的方法以及对应的R包/python模块有:
· ZINB-WaVE(R包: zinbwave)[7]
· scVI (python 模块: scvi)[8]
· DCA (python 模块: dca)[9]
regularized negative binomial regression (R包: sctransform; also being wrapped in Seurat) [10]。

与前面估算size factor的方法不同,这类模型拟合类方法通常将批次矫正和数据标准化结合到了一起,不需要分步处理。
Seurat: log-normalization vs. sctransform
或许大家会注意到R包Seurat提供了两种标准化的选择:log-normalization 和sctransform。Hafemeister et al.,[10] 对比了这两种方法,发现log-normalization对不同表达量的基因标准化效果不一致,只有中等以及低表达的基因被有效的标准化了,表明“size factor”并不是对所有基因都有效,并且这个是否有效的差异与测序深度相关 (Figure 1D from [10])。

而在r包sctransform中,他们通过构建regularized negative binomial regression 模型,对比发现模型残差能有效的标准化表达值,而且残差的方差(variance of residuals) 不受测序深度影响 (Figure 3C from [10])。在他们的pipeline中也提到,如果研究涉及到多个不同scran-seq数据的合并 (特别是不同protocols生成的数据),建议使用sctransform,运行时间比传统的log-normalization会短很多。

小tip: 数据标准化(normalization) 和批次矫正(batch correction) 之间有什么区别吗?标准化只考虑技术偏差,与有没有批次效应无关;而批次矫正,顾名思义,是特指出现在不同批次之间的差异,需要同时考虑技术偏差和生物学差异。技术偏差一般对具有相似特征 (例如长度,GC content) 的基因造成的影响也是类似的,而批次之间的生物学差异则要复杂的多,而且难以预估。因此这两个步骤涉及了不同的方法,大家千万主要不要混淆了这两个概念。
小编总结
标准化方法的选择很多,因为毕竟没有一种方法能适用于所有类型的scRNA-seq数据。在大多数的单细胞分析教程中log-normalization还是比较常用的方法,因为它相对简单并且容易实现。从我个人的分析经验来说,我尝试过用不同的数据对比文中提到的两大类方法,在细胞聚类上结果并没有显著的差异。不过因为具体数据的特异性,建议大家在了解方法的基础上,多多尝试不同的方法,特别在当聚类结果不太理想的时候。我们之后也会为大家介绍系统比较这些方法的文章,V信搜索:作图丫,可获取更多精彩内容。
参考资料/文献
1. Hwang, B., Lee, J.H. & Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med 50, 96 (2018). https://doi.org/10.1038/s12276-018-0071-8
2. Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol. 2019;15(6):e8746. Published 2019 Jun 19. doi:10.15252/msb.20188746
3. Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888-1902.e21. doi:10.1016/j.cell.2019.05.031
4. McCarthy DJ, Campbell KR, Lun ATL, Willis QF (2017). “Scater: pre-processing, quality control, normalisation and visualisation of single-cell RNA-seq data in R.” Bioinformatics, 33, 1179-1186. doi: 10.1093/bioinformatics/btw777.
5. Lun ATL, McCarthy DJ, Marioni JC (2016). “A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor.” F1000Res., 5, 2122. doi: 10.12688/f1000research.9501.2.
6. Risso D, Cole M (2020). scRNAseq: Collection of Public Single-Cell RNA-Seq Datasets. R package version 2.2.0.
7. Risso D, Perraudeau F, Gribkova S, Dudoit S, Vert J (2018). “A general and flexible method for signal extraction from single-cell RNA-seq data.” Nature Communications, 9, 284. https://doi.org/10.1038/s41467-017-02554-5.
8. Lopez R, Regier J, Cole MB, Jordan MI, Yosef N. Deep generative modeling for single-cell transcriptomics. Nat Methods. 2018;15(12):1053-1058. doi:10.1038/s41592-018-0229-2
9. Eraslan, G., Simon, L.M., Mircea, M. et al. Single-cell RNA-seq denoising using a deep count autoencoder. Nat Commun 10, 390 (2019). https://doi.org/10.1038/s41467-018-07931-2
10. Hafemeister, C., Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296 (2019). https://doi.org/10.1186/s13059-019-1874-1