前言——主要内容
这篇笔记是StatQuest系列笔记的第63节,这篇笔记跳过59节,60节,61节主要是因为第63节的内容是讲RPKM,FPKM和TPM这几个概念的区别,在进行差异分析前,有必要了解一下这几个概念。
测序的度量单位
在RNA-Seq中,我们通常使用RPKM(全称为Reads Per Kilobase Million)或FPKM(Fragments Per Kilobase Million)来进行均一化,这些均一化的数据(normalized data)主要解决两个问题:
- 测序深度(这个是指Million这一部分,侧重于数量),我们知道,测序深度越高,那么比对到基因上的reads数就越多;
- 基因的长度(这个指的是Kilobase这一部分,侧生于长度),我们知道,一个基因越长,那么比对到这个基因上的reads数就越多。
不过现在我们还使用TPM(全称为Transcripts per million)来作为均一化后的基因的表达单位,如下所示:

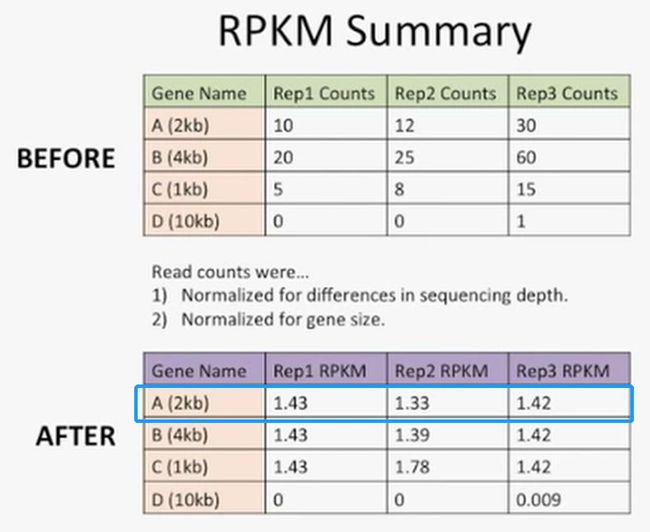
为了能够更好的区分这三个单位,即RPKM,FPKM和TPM,我们就通过一个简单的案例来说明一下,在这个案例中,我们一共有3个重复,分别为Rep1,Rep2和Rep3,检测了4个基因,分别为A,B,C,D,如下所示:
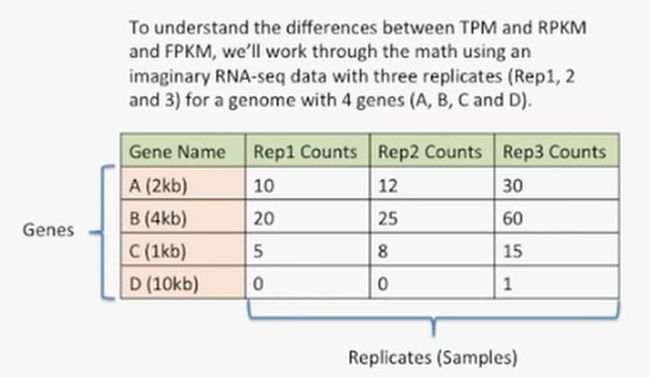
从上面的图表可以发现,Rep3的reads数比其它的2个更多,如下所示:

我们再看一下基因B,它的长度(4kb)是基因A(2kb)的2倍,这个或许就可以说明为什么基因B的reads数是基因A的reads数的2倍了,如下所示:

RPKM
RPKM的全称为Reads Per Kilobase Million,中文翻译就是每千个碱基的转录每百万映射读取的reads数。
现在我们使用RPKM来均一化这些数据,如下所示
均一化的步骤如下所示
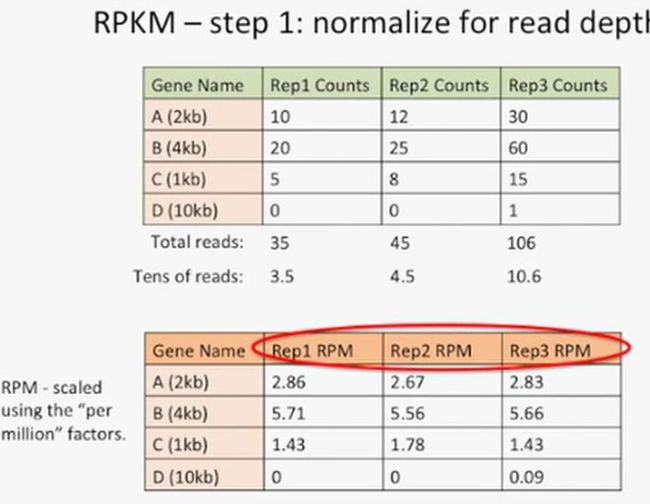
第一步:均一化read深度。
我们计算一下某个重复的总reads数,然后用它们除以10,这里我们只是简单地模拟一下,真正的应该是除以1000000,因为RPKM的M指的就是million,是百万,但如果要除以1百万的话,小数点太多,不太直观,如下所示:

现在就得到了总reads数除以10的结果,分别为3.5,4.5,10.6,此时,再用每个基因对应的reads数除以相应的前面结果,例如对于Rep1中的基因A来说,就是使用10除以3.5,也就是2.86,得到的这个单位我们称之为RPM,也就是reads per million,如下所示:
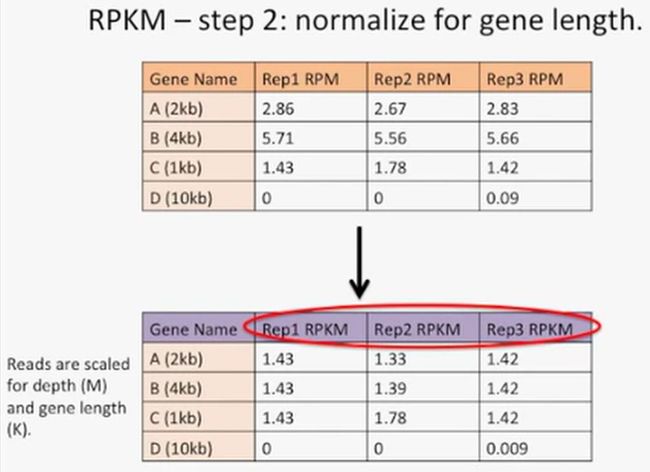
第二步:均一化基因长度。
在这一步骤中,我们需要对基因的长度进行均一化,如下所示:

此时,用RPM的数值除以每个基因的长度,例如,对于Rep1的基因A来说,就是2.86,基因A的长度是2kb,那么这个数值就是2.86除以2等于1.43,这就是RPKM,如下所示:
现在我们看一下RPKM的效果,在均一化之前的数据与均一化之后的数据,我们可以发现,经过均一化后的数据,每个重复里的每个基因的RPKM就非常接近了,如下所示:
FPKM
FPKM的全称为Fragments Per Kilobase Million,对应的中文就是每千个碱基的转录每百万映射读取的fragments。FPKM与RPKM的计算非常接近,其中区别就在于一个是FPKM的Fragemnts,而RPKM则是reads。
另外,RPKM通常用于单端测序,FPKM常用于双端测序,如下所示:

在RNA-Seq中,我们建库时会把DNA打断成小片段(fragment),两端加上接头,然后测序。
如果是单端测序,那么一个fragmetns就对应了一条read,如下所示:
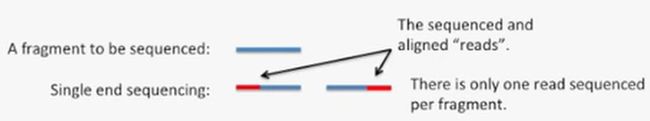
如果是双端测序,那么一条fragments就对应两条reads,当然,有时候双端测序也有可能出现一条fragment对应一条read(另外一条read有可能会因为质量低而被剔除),FPKM就保证了,一条fragment的两条reads不会被统计2次,如下所示:

也就是说FPKM是以fragment为准,而不是以reads数为准,它们的计算方式是一样的。
TPM
TPM的全称为Transcripts per million,中文就是每百万条reads的转录本。我们现在还以原来的案例,看一下TPM是如何计算的。
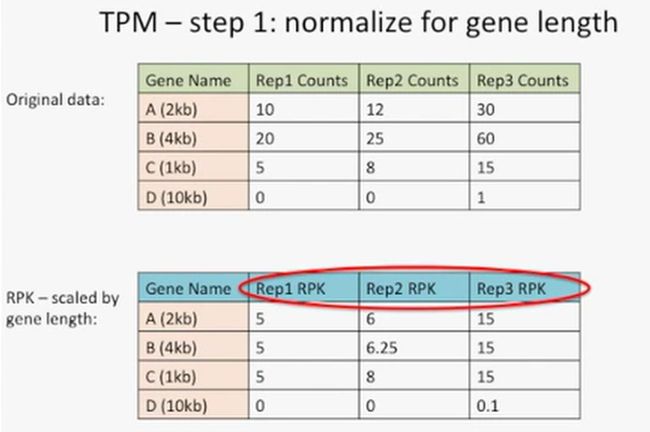
第一步:均一化基因长度。
每个重复中的每个基因的reads数除以该基因的长度,对于Rep1的基因A来说,就是10除以2,也就是5,也叫RPK,如下所示:
第二步:均一化测序深度。
先计算出一个重复中总的RPK之和,然后除以10,对于Rep1来说,总的RPK是15,除以10就是1.5,如下所示:
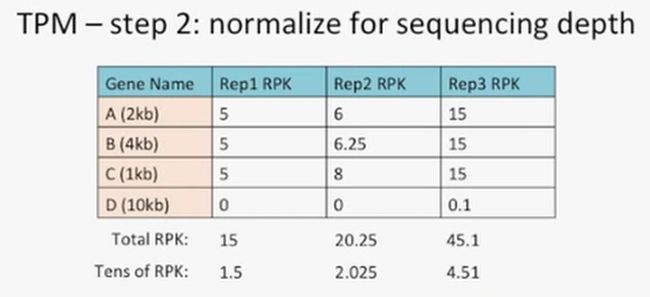
然后再用每个基因的RPK除以上面计算出来的数值,对于Rep1的基因A来说,就是5除以1.5,即3.33,如下所示:
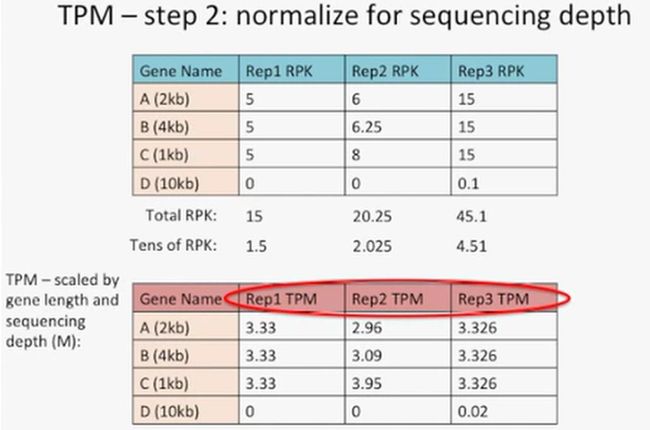
经过TPM这样的均一化后,我们可以发现,这3个重复的数值就比较接近了。
TPM和RPKM的区别
我们比较一下RPKM与TPM均一化扣的结果,如下所示:
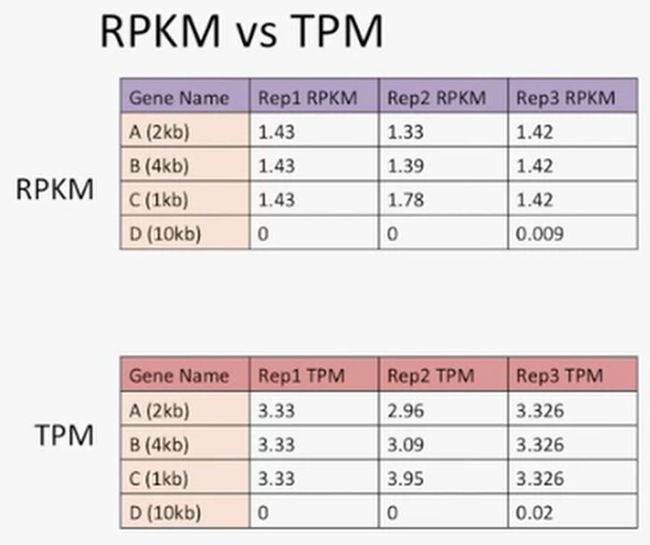
TPM和RPKM(FPKM)都是基于基因的长度与测序深度进行均一化的,但是它们也有所不同。我们看一下,对于每个重复来说,它们的总TPM和RPKM的数据数据之间的是存在一定差异的,如下所示:

对于RPKM来说,每个重复的总RPKM值不一样,分别是4.29,4.5和4.25。
对于TPM来说,每个重复的总TPM是相同的,都是10(也就是说总的TPM)。
我们再来看一下这两种的差异有何重要意义。
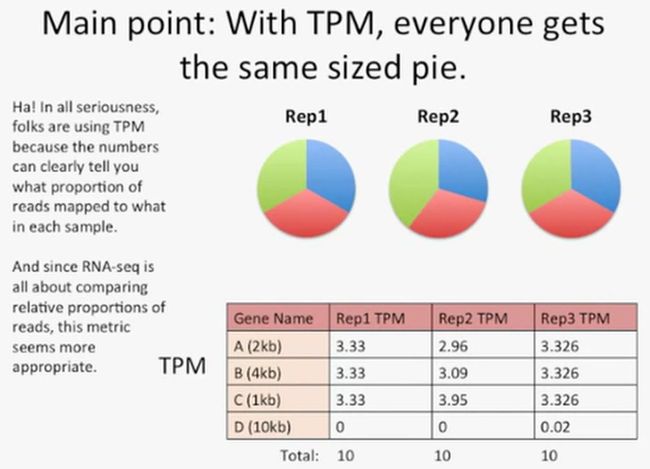
我们先看下面的3个饼图,每个饼图的有大小相同,数值都是10。从中我们可以发现,对于同样的饼图来说,一份3.33的扇形(它代表了Rep1中的基因A)肯定比一份3.32的扇形(它代表了Rep2中的基因A)要大。而TPM就能发现这种3.33和3.32的这种差异,我们可以看到,在Rep1的总reads数中,比对到基因A上的reads数的比例肯定比Rep3中相应的比例要高,如下所示:
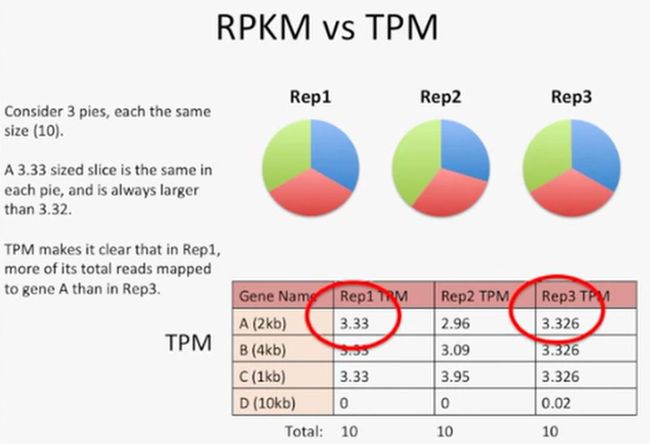
为什么会出现这种差异,我们可以再看上图,我们可以发现,在Rep3中,比对到基因D上的reads数所占的比例要大于Rep1中,比对到基因D的reads数的比例。通过TPM我们更容易发现,在每个重复中,比对到哪些基因上的reads更高或更低。
而对于RPKM这种均一化来说,很难比较比对到哪个基因上的总reads高,因为每个重复的总reads数都不同,也就是说这个饼图形的大小都不一样,如下所示:
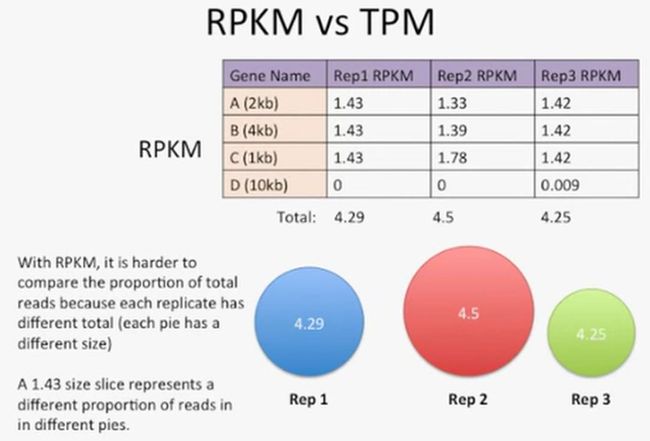
还看上图,对于Rep1中的基因A来说,它的RPKM是1.43,而在Rep3中,基因A的RPKM是1.42,那么我们不能直接说在Rep1中,比对到基因A上的总Reads数大于Rep3中基因A的总Reads数,也就是无法直接说在Rep1中基因A的转录水平高于Rep3中基因A的转录水平,因为Rep1和Rep3的总RPKM不同。
而对于TPM就不一样,每个重复的总TPM是一样的,这样很容易比较相同基因在不同的重复中的转录水平高低。由于RNA-Seq主要是研究基因的转录水平,也就是说研究基因的转录本的reads的相对比例,因此使用TPM更加合理,如下所示:
CPM
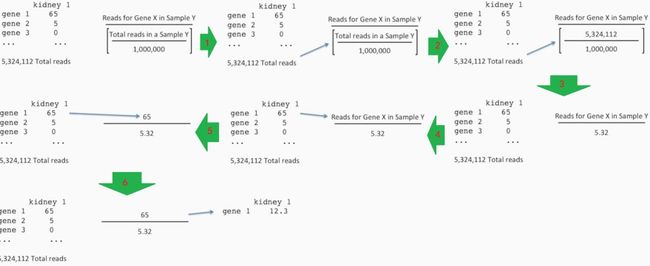
有的时候还需要做到CPM,CPM的计算流程为:①总reads数除以100万;②用每个样本中的每个基因对应的reads数再除以①中的数字,就是CPM。
总reads数除以100万主要是为了方便计算CPM,否则CPM的数值会非常小,不方便,如下所示:
用途:在某些情况下,只想了解每个基因被覆盖到的相对reads数,而不希望对其做长度校正,就会使用这个指标。在某些RNA-seq文章或一些软件输出结果中(如edgeR)会出现。CPM只对read count相对总reads数做了数量的均一化。当如果想进行表达量的基因间比较,则就需要考虑基因长度的不同。如果进一步做长度的均一化,就是RPKM。