刘小泽写于2020.6.27
为何取名叫“交响乐”?因为单细胞分析就像一个大乐团,需要各个流程的协同配合
单细胞交响乐1-常用的数据结构SingleCellExperiment
单细胞交响乐2-scRNAseq从实验到下游简介
单细胞交响乐3-scRNA的细胞质控
1 前言
scRNA数据中文库的差异还是很常见的,来源于:不同细胞的起始底物浓度不同,导致cDNA捕获或PCR扩增效率差异。归一化的目的就是去除细胞间与真实表达量无关的技术因素,方便后续比较。
这里需要说明:归一化与批次处理还是不同的。归一化不管实验的批次因素,只考虑细胞中存在的技术误差(比如测序深度),而批次处理既要考虑实验批次,又要考虑技术误差(比如不同实验时间、不同细胞系、不同文库制备方法、不同测序方法、不同测序深度)。技术误差对不同基因的影响是相似的,比如有的技术误差会影响基因长度、GC含量,那么基本上所有基因都会受影响。但不同的批次产生的影响是不同的,比如不同时间做的实验效果就是不一样,那么基因的表达量可能也会受到这个影响。尽管有的R包可以同时处理技术误差和批次效应(例如zinbwave),但大部分的分析还是各顾各的。
因此,这里只需要记得:归一化,归的是技术误差,而不是批次效应即可
原文中提到:We will mostly focus our attention on scaling normalization, which is the simplest and most commonly used class of normalization strategies.
个人比较喜欢叫normalization为归一化,scale为标准化【只是一种称呼习惯而已】。
当然也有朋友喜欢叫normalization为标准化。其实,scale与normalization的定义也不用掰扯太清楚,谁包含谁不用区分太明白。只需要知道分别做了什么事就可以了,一般来说,应该先进行normalization,再进行scale,而scale的结果会用来后续的降维和聚类
- scale:This involves dividing all counts for each cell by a cell-specific scaling factor, often called a “size factor” (Anders and Huber 2010)
The size factor for each cell represents the estimate of the relative bias in that cell, so division of its counts by its size factor should remove that bias.- Normalization "normalizes" within the cell for the difference in sequenicng depth / mRNA throughput. 主要着眼于样本的文库大小差异
Scaling "normalizes" across the sample for differences in range of variation of expression of genes . 主要着眼于基因的表达分布差异- 在Seurat中,一般得到原始表达矩阵并过滤后,会进行
NormalizeData(),当然也可以使用自己的归一化结果(例如TPM结果,只是需要再log一下);在挑选HVGs之后,降维处理之前,还需要用到ScaleData()进行标准化。对每个进行center后的值再除以标准差(就是进行了一个z-score的操作)【详细探索可以看我之前写的:https://mp.weixin.qq.com/s/6ioR3JE0wKg6M-YAsLBcTA】
需要说明的是:本文的介绍基本都是基于Scater和Scran包,没有Seurat的影子,所以也看不到ScaleData的操作。大部分都是对文库差异进行的处理,集中体现在计算size factor,取log进行数据缩放,因此主要就是归一化处理,而没有看到z-score处理。这个也不用奇怪,每个包都有每个包的处理方法,也没有一个定论说就非要进行Seurat的scale处理
数据准备
数据依然给大家准备好了,sce.zeisel链接:https://share.weiyun.com/mNwJS8U9 密码:g8379h
library(scRNAseq)
# 数据大概18M
sce.zeisel <- ZeiselBrainData()
rownames(sce.zeisel)
# 会看到有一些奇怪的基因名
# 将每个细胞的所有基因表达量加起来
library(scater)
sce.zeisel <- aggregateAcrossFeatures(sce.zeisel,
id=sub("_loc[0-9]+$", "", rownames(sce.zeisel)))
# 基因注释
library(org.Mm.eg.db)
rowData(sce.zeisel)$Ensembl <- mapIds(org.Mm.eg.db,
keys=rownames(sce.zeisel), keytype="SYMBOL", column="ENSEMBL")
# 质控(先perCellQCMetrics,后quickPerCellQC)
stats <- perCellQCMetrics(sce.zeisel, subsets=list(
Mt=rowData(sce.zeisel)$featureType=="mito"))
qc <- quickPerCellQC(stats, percent_subsets=c("altexps_ERCC_percent",
"subsets_Mt_percent"))
sce.zeisel <- sce.zeisel[,!qc$discard]
sce.zeisel
## class: SingleCellExperiment
## dim: 19839 2816
## metadata(0):
## assays(1): counts
## rownames(19839): 0610005C13Rik 0610007N19Rik ... Zzef1 Zzz3
## rowData names(2): featureType Ensembl
## colnames(2816): 1772071015_C02 1772071017_G12 ... 1772063068_D01
## 1772066098_A12
## colData names(10): tissue group # ... level1class level2class
## reducedDimNames(0):
## altExpNames(2): ERCC repeat
2 各种Size Factors的计算
2.1 文库相关的size factor | library size normalization
这个的计算也是最简单最常用的
文库大小指的就是:每个细胞中所有的基因表达量之和
进行归一化时,需要根据每个细胞的文库大小各自计算一个”library size factor“,而这些factor的均值为1
lib.sf.zeisel <- librarySizeFactors(sce.zeisel)
# 说明是对列操作
> length(lib.sf.zeisel)
[1] 2731
> ncol(sce.zeisel)
[1] 2731
# size factor 均值为1
> summary(lib.sf.zeisel)
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.1725 0.5778 0.8701 1.0000 1.2716 4.0096
对size factor做个直方图
hist(log10(lib.sf.zeisel), xlab="Log10[Size factor]", col='grey80')
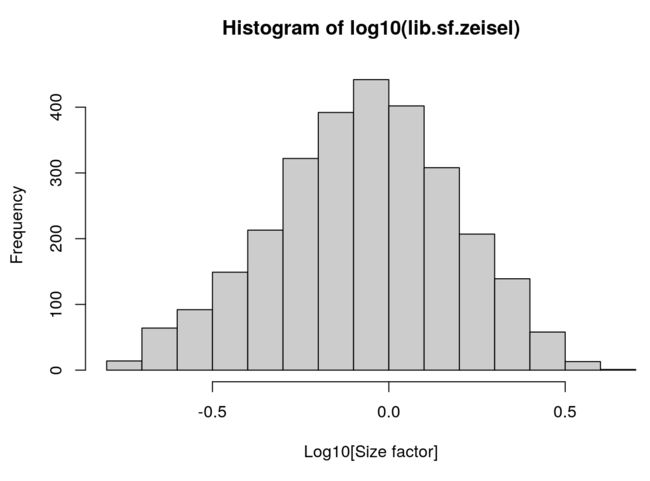
文库相关的size factor 基于的假设是:任意两对细胞间的差异表达基因都是平衡的。也就是说,一组基因的上调表达程度,势必会被另一组基因的下调表达程度相抵消。但是在scRNA中差异基因表达并不是平衡的(也称为:存在composition biases),因此library size normalization可能不会得到准确的归一化结果,仅仅是能用,但不是最精确。
不过话又说回来,精确的归一化结果也不是scRNA数据探索的主要任务。还是要记得,归一化的目的是更好地为下游展示数据做铺垫。因此使用library size normalization对细胞分群和鉴定每群的marker基因也是足够的。
2.2 去卷积方法计算size factor| Normalization by deconvolution
前面提到scRNA的数据存在composition biases,也就是样本间基因差异表达的不均衡性。
先来理解composition biases,现在想象一个例子:两个细胞A、B,一个基因X在A细胞相比于B细胞表达量更高。这种高表达意味着:
- 当细胞文库大小一定(例如实验采用library quantification/文库定量的方法),更多的测序资源偏向X基因,势必会降低其他本来不是差异表达基因的覆盖度【简言之,一个盘子就这么多水果,一个异类胃口大开,吃了很多,其他人本来也能吃饱,但现在也要挨饿】
- 当不限制细胞文库大小时,X基因的reads或UMIs增加,也会增加总体的文库大小,导致计算的library size factor增加,原本非差异基因的表达量也会由于size factor的增加而相对之前降低【简言之,一个异类拉高了整体平均成绩,导致其他本来成绩平平的学生看上去成绩发生了下降】
这两种情况都会导致一个结果:细胞A的其他非差异基因都会由于X基因的”过度表现“而不经意地”下调“
那么如何去除这种composition biases呢?之前在bulk RNA-Seq中研究非常透彻了。例如DESeq2使用的estimateSizeFactorsFromMatrix(),edgeR使用的calcNormFactors() 都在归一化过程中考虑到了这点。它的假设是:大部分基因都不是差异基因。
但是单细胞数据不能使用bulk 转录组的方法,因为存在大量的低表达和0表达量。根据Lun, Bach, and Marioni 2016的研究,单细胞可以这么操作:
- 先混合:Pool counts from many cells to increase the size of the counts
- 后去卷积:Pool-based size factors are then “deconvolved” into cell-based factors for normalization of each cell’s expression profile.
library(scran)
set.seed(100)
clust.zeisel <- quickCluster(sce.zeisel)
table(clust.zeisel)
# clust.zeisel
# 1 2 3 4 5 6 7 8 9 10 11 12
# 215 147 237 565 162 350 276 291 175 109 103 101
# 然后去卷积
deconv.sf.zeisel <- calculateSumFactors(sce.zeisel, cluster=clust.zeisel)
summary(deconv.sf.zeisel)
# Min. 1st Qu. Median Mean 3rd Qu. Max.
# 0.1292 0.4954 0.8337 1.0000 1.3145 4.4807
quickCluster使用了 irlba 的PCA方法来进行分群操作(注意这个分群和后面真正的分群操作不是一回事,这个顶多算是pre-clustering操作,目的就是给去卷积铺路),并且它每次运行结果都是随机的,因此需要设置随机种子来满足重复性。它得到的每群细胞都分别进行归一化,得到size factors。
# 看一下这个数据包含了这么多的细胞类型
> table(sce.zeisel$level1class)
astrocytes_ependymal endothelial-mural
160 132
interneurons microglia
290 67
oligodendrocytes pyramidal CA1
749 938
pyramidal SS
395
# 将去卷积的size factor与之前常规方法得到的size factor进行对比
plot(lib.sf.zeisel, deconv.sf.zeisel, xlab="Library size factor",
ylab="Deconvolution size factor", log='xy', pch=16,
col=as.integer(factor(sce.zeisel$level1class)))
abline(a=0, b=1, col="red") #截距为0,斜率为1
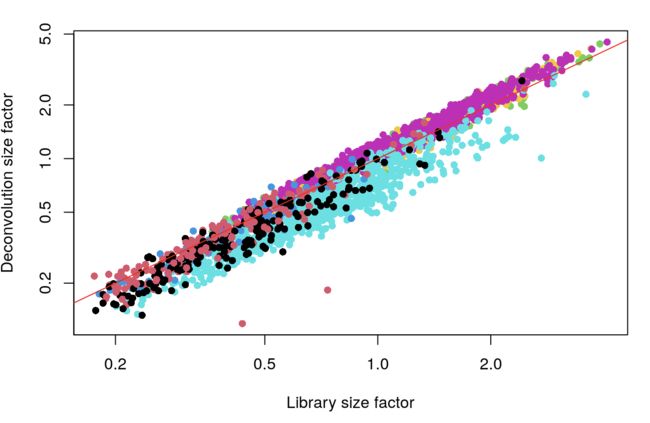
中间的红线上的点表示去卷积和常规方法得到的size factor相等;不同颜色表示不同的细胞类型。可以看到去卷积的方法的确能暴露出细胞类型对文库大小的影响,也证明了composition biases的确存在,并且在不同的细胞类型之间还很显著。利用去卷积的size factor会得到更准确的结果,因为它考虑的问题更全面。
上面也说过,使用library size normalization对细胞分群和鉴定每群的marker基因是足够的。这里使用去卷积得到更准确的结果,虽然对分群改善不大,但对于每个基因的表达量数据估计与解释还是很重要的,因为它考虑了composition biases的影响。
2.3 使用spike-in计算size factor
它基于的假设是:外源RNA(spike-in)添加到每个细胞时的量都是一样的。如果出现同一spike-in转录本表达量在各个细胞之间的差异,问题只能是细胞相关,例如不同细胞捕获效率、不同细胞测序深度等等。
因此,计算spike-in size factor的目的也正是去除这方面的偏差。相比之前的两种方法,spike-in的方法不再基于生物学背景假设(比如是否存在很多差异基因),而是假设:
- 添加到每个细胞的spike-in的量都是一定的
- 对偏差的反应,和内源基因是一样的
下面快速探索这个过程:
# 准备数据
library(scRNAseq)
sce.richard <- RichardTCellData()
sce.richard <- sce.richard[,sce.richard$`single cell quality`=="OK"]
sce.richard #看到有ERCC存在
## class: SingleCellExperiment
## dim: 46603 528
## metadata(0):
## assays(1): counts
## rownames(46603): ENSMUSG00000102693 ENSMUSG00000064842 ...
## ENSMUSG00000096730 ENSMUSG00000095742
## rowData names(0):
## colnames(528): SLX-12611.N701_S502. SLX-12611.N702_S502. ...
## SLX-12612.i712_i522. SLX-12612.i714_i522.
## colData names(13): age individual ... stimulus time
## reducedDimNames(0):
## altExpNames(1): ERCC
使用computeSpikeFactors() 计算所有细胞的spike-in size factor
sce.richard <- computeSpikeFactors(sce.richard, "ERCC")
summary(sizeFactors(sce.richard))
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.1247 0.4282 0.6274 1.0000 1.0699 23.3161
再做一个spike-in与去卷积结果的对比图:
to.plot <- data.frame(
DeconvFactor=calculateSumFactors(sce.richard),
SpikeFactor=sizeFactors(sce.richard),
Stimulus=sce.richard$stimulus,
Time=sce.richard$time
)
ggplot(to.plot, aes(x=DeconvFactor, y=SpikeFactor, color=Time)) +
geom_point() + facet_wrap(~Stimulus) + scale_x_log10() +
scale_y_log10() + geom_abline(intercept=0, slope=1, color="red")
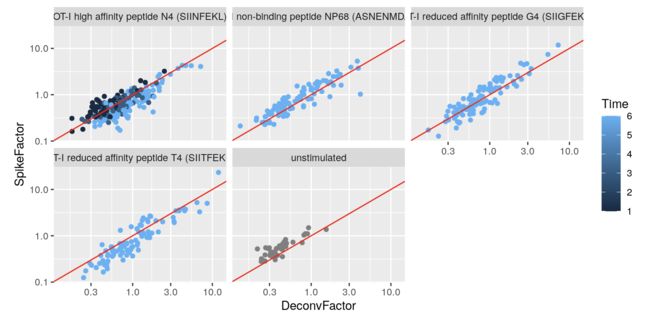
发现每个处理的spike-in size factor与去卷积的结果呈正相关,表示它们都捕获到了类似的技术偏差(测序深度、捕获效率)
同时注意到随着时间的增加(如左上图),spike-in factors在下降,去卷积factors在增加,可以解释为:随着时间的推移,生物合成活性在增加,总体RNA含量在增加,而spike-in的含量是一定的,因此每个文库中各个spike-in的比例减少(导致spike-in size factor结果减少),而总体文库增大(导致文库size factor增加)
小结
对于归一化方法的选择,取决于生物学假设。大多数情况下,总体RNA含量的变化我们并不感兴趣,可以直接用library size或deconvolution factors当做系统性偏差去除。但如果总体RNA含量变化与某个感兴趣的生物学过程相关,例如细胞周期活性或T细胞激活,spike-in的归一化方法就可以把这种变化保留下来,而去除其他系统性偏差。
另外,不管我们关不关心总体RNA含量,对于spike-in转录本的归一化都有特殊对待,不能使用针对内源基因的方法,比如可以利用modelGeneVarWithSpikes() 得到单独的spike-in相关的size factor
3 得到Size Factors后进行数据转换
3.1 logNormCounts
log-transformation这种是最常用、最简单、最易理解的方法
一旦得到了size factors,就可以利用scater包的logNormCounts()函数计算每个细胞归一化后的结果。计算也很简单,就是用某个细胞中每个基因或spike-in转录本的表达量除以这个细胞计算的size factor,最后还进行一个log转换,得到一个新的矩阵:logcounts 【不过这个名字并不是很贴切,只是因为拼写简单,真实应该是:log-transformed normalized expression values。而不是单纯字面意思取个log】
set.seed(100)
# 这里一看有pre-clustering就知道是利用的去卷积的size factor
clust.zeisel <- quickCluster(sce.zeisel)
sce.zeisel <- computeSumFactors(sce.zeisel, cluster=clust.zeisel, min.mean=0.1)
sce.zeisel <- logNormCounts(sce.zeisel)
# 最后的结果多了一项
assayNames(sce.zeisel)
## [1] "counts" "logcounts"
至于为什么取log? 就是为了不受真实值的影响,而关注变化倍数。这里举个例子,X基因在细胞A的表达量是50,在B细胞的表达量是10;而Y基因在细胞A的表达量是1000,在B细胞的表达量是1100。哪个基因更能吸引你的注意呢?我们是不是会关注变化的倍数?
另外,既然要取log,就应该要注意存在很多0表达量的数值,因此log前还需要给表达量加上1,log1p = log(x + 1),这个1就称作:pseudo-count。
注意,每个包的log-transforming方法存在差异:
scran和scater使用的一样,是logNormCounts,用表达量除以这个细胞计算的size factor;
但Seurat使用的LogNormalize计算方法是:normalizes the feature expression measurements for each cell by the total expression, multiplies this by a scale factor (10,000 by default), and log-transforms the result,翻译成公式就是:log1p(value/colSums[cell-idx] *scale_factor)
3.2 其他方法
其实默认大家都在用log-transformation方法,只是有时还有更特殊的需求
logNormCounts还有一个参数:downsample=T ,当使用常规流程做完后发现PCA分群的结果可能受到size factor的影响(即使之前通过归一化试图去除文库大小的影响)。既然文库大小影响还在,那么就试图减弱这种影响,于是想到减少样本数量,用更少的样本再重新归一化【但这种情况非常特殊】
图中看到选取更少的样本,结果变得清楚许多,size factor的影响比之前小了很多。于是可以推断,之前PCA的结果很可能是还存在技术偏差导致的文库差异

另外还有更复杂的方法可以看:sctransform 、DESeq2,使用了variance stabilizing transformations方法
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
