旧号无故被封,小号再发一次
更多空间转录组文章:
1. 新版10X Visium
- 【10X空间转录组Visium】(一)Space Ranger 1.0.0(更新于20191205)
- 【10X空间转录组Visium】(二)Loupe Browser 4.0.0
- 【10X空间转录组Visium】(三)跑通Visium全流程记录
- 【10X空间转录组Visium】(四)R下游分析的探索性代码示例
- 【10X空间转录组Visium】(五)Visium原理、流程与产品
- 【10X空间转录组Visium】(六)新版Seurat v3.2分析Visium空间转录组结果的代码实操
- 【10X空间转录组Visium】(七)思考新版Seurat V3.2作者在Github给予的回答
2. 旧版Sptial
- 【旧版空间转录组Spatial】(一)ST Spot Detector使用指南
- 【旧版空间转录组Spatial】(二)跑通流程试验记录
- 【旧版空间转录组Spatial】(三)ST Spot Detector实操记录
环境说明:
为了避免与之前的Seurat包冲突,当前环境没有安装之前的Seurat版本的win10 Rstudio,而非服务器生产环境
一、Seurat v3.2 对空间转录组Visium的结果分析中实现的功能:
- 归一化
- 降维和聚类
- 检测空间可变特征(spatially-variable features)
- 互动式的可视化
- 与单细胞RNA-seq数据整合
- 处理多个切片
二、安装Seurat v3.2 :
#For learning Seurat v2.3
#Author:Robin 2019.12.22
# Enter commands in R (or R studio, if installed)
# Install the devtools package from Hadley Wickham
install.packages("devtools")
devtools::install_github("satijalab/seurat", ref = "spatial")
devtools::install_github('satijalab/seurat-data')
一开始尝试了几次都装不成功,后面突然就行了,而且网速还很快,果然是看运气的。。
成功安装!
> library(Seurat)
> package.version("Seurat")
[1] "3.1.2" #说是官网v3.2,实际上是v3.1.2
加载包:
library(Seurat)
library(SeuratData)
library(ggplot2)
library(cowplot)
library(dplyr)
对于第一个小插图,我们分析了使用10x Genomics 的Visium技术生成的数据集。我们将扩展Seurat以便在不久的将来使用其他数据类型,包括SLIDE-Seq,STARmap和MERFISH。
三、下载数据集(但一般不这么做)
在这里,我们将使用使用Visium v1化学方法生成的最近发布的矢状小鼠大脑切片数据集。小鼠大脑的矢状面前段后段(There are two serial anterior sections, and two (matched) serial posterior sections.)
- 这将读取spaceranger管道的输出,并返回一个Seurat对象,该对象包含spot-level表达数据以及组织切片的关联图像。
- 您还可以使用我们的SeuratData包来轻松访问数据,如下所示。安装数据集后,您可以键入
?stxBrain以了解更多信息。
InstallData("stxBrain")
brain <- LoadData("stxBrain", type = "anterior1")
由于国内网速很慢,建议直接在此处下载数据包到本地,然后使用Load10X_Spatial函数将其加载到Seurat中。
四、加载10X Visiumspaceranger管道中生成的结果文件
在生产中,我们实际上加载的数据集通常是自己经过Visium spaceranger管道生成的结果文件,而非直接从网上下载的数据集。
从10X Visium流程中生成结果文件的方法: 10X空间转录组Visium学习笔记(三)跑通Visium全流程记录
查看目录结构
$cd /V1_Adult_Mouse_Brain/outs
$tree -L 2
.
├── analysis
│ ├── clustering
│ ├── diffexp
│ ├── pca
│ ├── tsne
│ └── umap
├── cloupe.cloupe
├── filtered_feature_bc_matrix
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── filtered_feature_bc_matrix.h5 #实际上需要的文件①
├── metrics_summary.csv
├── molecule_info.h5
├── possorted_genome_bam.bam
├── possorted_genome_bam.bam.bai
├── raw_feature_bc_matrix
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── raw_feature_bc_matrix.h5
├── spatial # 实际上需要的文件②;记录空间信息:用户提供的原始全分辨率brightfield图像的下采样版本。下采样是通过box滤波实现的,它对全分辨率图像中像素块的RGB值进行平均,得到下采样图像中一个像素点的RGB值。
│ ├── aligned_fiducials.jpg
│ ├── detected_tissue_image.jpg
│ ├── scalefactors_json.json
│ ├── tissue_hires_image.png
│ ├── tissue_lowres_image.png
│ └── tissue_positions_list.csv
└── web_summary.html
9 directories, 20 files
加载数据
此处是将服务器之前跑的Space Ranger的输出结果 filtered_feature_bc_matrix.h5文件和一个spatial文件夹放置到本地win10环境中test/目录下
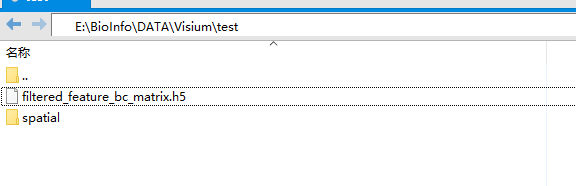
读取文件需要安装好
hdf5r包
> brain<-Load10X_Spatial(
+ data.dir = 'E:\\BioInfo\\DATA\\Visium\\test', #该目录包含10X提供的matrix.mtx,genes.tsv(或features.tsv)和barcodes.tsv文件。
+ assay = "Spatial",
+ slice = "slice1", #组织切片存储图像的名称
+ filter.matrix = TRUE, #仅保留已确定位于组织上方的spot
+ to.upper = FALSE, #将所有功能名称转换为大写。 例如,当分析需要在人类和小鼠基因名称之间进行比较时,该功能将非常有用。
+ )
# 查看Seurat对象信息
> brain
An object of class Seurat
31053 features across 2698 samples within 1 assay
Active assay: Spatial (31053 features)
当然,也可以?stxBrain会给出类似10X ATAC构建Seurat对象那种方法,不过可能有些麻烦:Seurat 新版教程:分析空间转录组数据
Seurat中如何存储空间数据?
来自10x的visium数据包含以下数据类型:
- 基因表达矩阵的点
- 组织切片的图像(从数据采集期间的H&E染色获得)
- 将原始高分辨率图像与此处用于可视化的较低分辨率图像相关联的比例因子
- 在Seurat对象中,基因表达矩阵的spot类似于典型的“ RNA”
Assay,但它包含spot水平,而不是单细胞水平数据。 - 图像本身存储在Seurat对象的
images新插槽中。 - 所述
images槽图还存储必要的信息,以将spot与其在组织图像上的物理位置相关联
 Seurat对象结构跟以前差不多
Seurat对象结构跟以前差不多
五、数据预处理
1. 数据标准化 Normalization
与scRNA-seq实验相似。除了测序深度的差异,对于空间数据集,分子计数/spot的差异可能很大,尤其是如果整个组织的细胞密度存在差异时。
plot1 <- VlnPlot(brain, features = "nCount_Spatial", pt.size = 0.5) + NoLegend()
plot2 <- SpatialFeaturePlot(brain, features = "nCount_Spatial") + theme(legend.position = "right")
plot_grid(plot1, plot2)

- 这些图表明,分子计数(molecular counts)在点间的差异不仅是技术上的,而且还取决于组织的解剖结构(组织中神经元耗竭的区域(如皮层白质),可重复显示较低的分子数)
- 因此,标准方法(如LogNormalize函数)可能会有问题,因为它会强制每个数据点在标准化之后具有相同的底层“大小”underlying ‘size’
- 作为一种替代方法,建议使用
sctransform(Hafemeister和Satija,已出版),它构建了基因表达的正则化负二项模型,以便在保留生物学差异的同时考虑技术伪像。
有关sctransform的更多信息,请参见 here的预印和here的Seurat教程。sctransform将数据归一化,检测高方差特征high-variance features,并将数据存储在SCTassay中。
brain <- SCTransform(brain, assay = "Spatial", verbose = FALSE)
- sct 与 log-normalization的比较
为了探索规范化方法的差异,我们检查了sctransform和log规范化结果如何与UMI数量相关。
为了进行比较,我们首先重新运行sctransform以存储所有基因的值,然后通过NormalizeData进行 log-normalization 过程。
# rerun normalization to store sctransform residuals for all genes
brain <- SCTransform(brain, assay = "Spatial", return.only.var.genes = FALSE, verbose = FALSE)
# also run standard log normalization for comparison
brain <- NormalizeData(brain, verbose = FALSE, assay = "Spatial") #这步有些慢
第一次运行NormalizeData的报错:"Error: std::bad_alloc".
解决方法:可能是内存不够,关掉了没用的网页,重新打开Rstudio重新运行,问题解决。
# Computes the correlation of the log normalized data and sctransform residuals with the number of UMIs
brain <- GroupCorrelation(brain, group.assay = "Spatial", assay = "Spatial", slot = "data", do.plot = FALSE)
brain <- GroupCorrelation(brain, group.assay = "Spatial", assay = "SCT", slot = "scale.data", do.plot = FALSE)
p1 <- GroupCorrelationPlot(brain, assay = "Spatial", cor = "nCount_Spatial_cor") + ggtitle("Log Normalization") +
theme(plot.title = element_text(hjust = 0.5))
p2 <- GroupCorrelationPlot(brain, assay = "SCT", cor = "nCount_Spatial_cor") + ggtitle("SCTransform Normalization") +
theme(plot.title = element_text(hjust = 0.5))
p3 <- plot_grid(p1, p2)
ggsave(p3, filename = "lOG_VS_SCT.png")

- 对于上面的箱线图,我们计算每个特征(基因)与UMI数量(此处为变量
nCount_Spatial)的相关性。然后,我们根据基因的平均表达量将基因分组,并生成这些相关系数的箱线图。 - 您可以看到 log-normalization 未能使前三组中的基因充分归一化,这表明技术因素继续影响高表达的基因的归一化表达估计值。相反,sctransform归一化实质上减轻了这种影响。
六、基因表达可视化
在Seurat v3.2中,我们加入了新的功能来探索和与空间数据固有的可视化特性。
Seurat的SpatialFeaturePlot函数扩展了FeaturePlot, 可以将表达数据覆盖在组织组织上。例如,在这个小鼠大脑数据中,Hpca基因是一个强烈的海马区(hippocampal )marker ,Ttr是一个脉络丛(choroid plexus)marker。
SpatialFeaturePlot(brain, features = c("Hpca", "Ttr"))
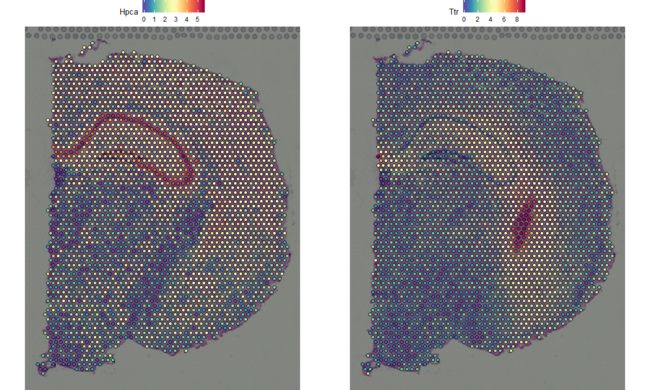
Seurat中的默认参数强调分子数据的可视化。但是,您还可以通过更改以下参数来调整斑点的大小(及其透明度),以改善组织学图像的可视化:
-
pt.size.factor-这将缩放斑点的大小。默认值为1.6 -
alpha-最小和最大透明度。默认值为c(1,1)。 - 尝试设置为
alphac(0.1,1),以降低具有较低表达的点的透明度
p1 <- SpatialFeaturePlot(brain, features = "Ttr", pt.size.factor = 1)
p2 <- SpatialFeaturePlot(brain, features = "Ttr", alpha = c(0.1, 1))
plot_grid(p1, p2)

七、降维,聚类和可视化
然后,我们可以使用与scRNA-seq分析相同的工作流程,对RNA表达数据进行降维和聚类。
brain <- RunPCA(brain, assay = "SCT", verbose = FALSE) #选择SCT归一化的assay
brain <- FindNeighbors(brain, reduction = "pca", dims = 1:30)
brain <- FindClusters(brain, verbose = FALSE)
brain <- RunUMAP(brain, reduction = "pca", dims = 1:30)
再然后,我们可以在UMAP空间(使用DimPlot)或使用SpatialDimPlot将聚类clustering的结果显示在图像上
p1 <- DimPlot(brain, reduction = "umap", label = TRUE)
p2 <- SpatialDimPlot(brain, label = TRUE, label.size = 3)
plot_grid(p1, p2)
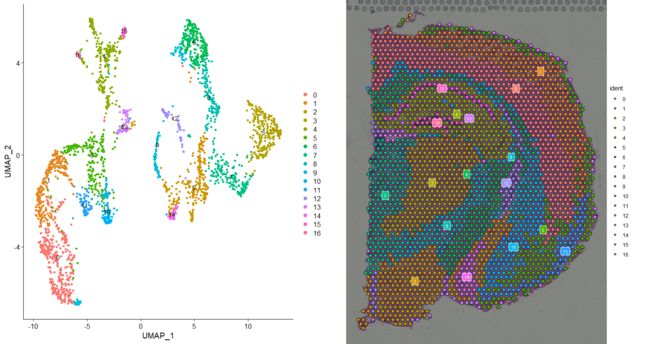
由于颜色太多,因此可视化哪个立体像素voxel属于哪个cluster可能是一个挑战。
解决方案:
- 设置
label参数会在每个cluster的中间处放置一个彩色框(请参见上图), - 而SpatialDimPlot的
do.hover参数允许交互式地查看当前点的标识。
# move your mouse
SpatialDimPlot(brain, do.hover = TRUE)
您还可以在SpatialDimPlot上使用cells.highlight参数在上划分特定的关注细胞,这对于区分单个cluster的空间定位非常有用。如下所示:
SpatialDimPlot(brain, cells.highlight = CellsByIdentities(object = brain, idents = c(1, 2, 5, 3,
4, 8)), facet.highlight = TRUE, ncol = 3)
此外,
LinkedDimPlot和LinkedFeaturePlot函数支持交互式可视化。这些图将UMAP表示与组织图像表示联系起来,并允许交互选择。例如,您可以在UMAP图中选择一个区域,图像表示中相应的点将突出显示。
LinkedDimPlot(brain)
八、识别空间可变特征
Seurat提供了两种工作流程来识别与组织内空间位置相关的分子特征。
- 第一种方法,是基于组织内预先标注的解剖区域执行差异表达,这可以从无监督聚类或先验知识中确定。在这种情况下,此策略将起作用,因为上面的cluster显示出明显的空间限制。
de_markers <- FindMarkers(brain, ident.1 = 0, ident.2 = 1)
SpatialFeaturePlot(object = brain, features = rownames(de_markers)[1:3], alpha = c(0.1, 1), ncol = 3)
-
第二种方法,
FindSpatiallyVariables在中实施的是在没有预先注释的情况下搜索展现出空间模式spatial patterning的特征。默认参数(
method = 'markvariogram)受Trendsceek启发,该工具将空间转录组学数据建模为标记点过程(mark point process),并计算“variogram”,以识别其表达水平取决于其空间位置的基因。更具体地说,此过程计算 gamma(r) 值,该值测量相距某个“ r”距离的两个点之间的依赖性。默认情况下,我们在这些分析中使用 r-value为“ 5”,并且仅计算可变基因的这些值(变异variation的计算独立于空间位置)以节省时间。
文献中有多种方法可以完成此类任务,包括SpatialDE和Splotch。Seurat团队鼓励感兴趣的用户探索这些方法,并希望在不久的将来为其提供支持。
现在,我们可视化此方法确定的前6个特征的表达情况。
top.features <- head(SpatiallyVariableFeatures(brain, selection.method = "markvariogram"), 6)
SpatialFeaturePlot(brain, features = top.features, ncol = 3, alpha = c(0.1, 1))
这步可能有点久

九、可视化解剖区域中取子集(Subset out)
与单细胞对象一样,您可以对对象进行取子集操作,以便重点集中在处理数据的子集。在这里,我们大概将额叶皮层(frontal cortex)进行取子集操作。此过程还有助于在下一节中将这些数据与皮质(cortical )scRNA-seq数据集整合。
PS:取得子集这步目前还是有些坑
具体请看:https://www.jianshu.com/p/497efa6b727f

所以这里往下的我都是用官网的结果,还没有尝试自己跑
- 首先,我们获取clusters的子集
cortex <- subset(brain, idents = c(1, 2, 3, 5, 6, 7))
- 然后根据精确位置进一步细分。
# now remove additional cells, use SpatialDimPlots to visualize what to remove
# SpatialDimPlot(cortex,cells.highlight = WhichCells(cortex, expression = image_imagerow > 400 |
# image_imagecol < 150))
cortex <- subset(cortex, anterior1_imagerow > 400 | anterior1_imagecol < 150, invert = TRUE)
cortex <- subset(cortex, anterior1_imagerow > 275 & anterior1_imagecol > 370, invert = TRUE)
cortex <- subset(cortex, anterior1_imagerow > 250 & anterior1_imagecol > 440, invert = TRUE)
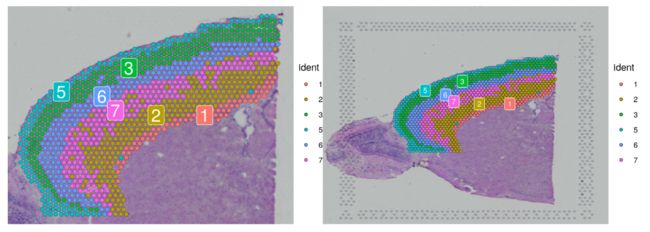
- 取子集后,我们可以在完整图像或裁剪图像上对皮质细胞进行可视化。
p1 <- SpatialDimPlot(cortex, crop = TRUE, label = TRUE)
p2 <- SpatialDimPlot(cortex, crop = FALSE, label = TRUE, pt.size.factor = 1, label.size = 3)
plot_grid(p1, p2)
十、与单细胞数据整合进行联合分析(10X一贯的玩法)
- 在〜50um时,来自于visium测定的 spots 将涵盖多个细胞的表达谱。对于可获得scRNA-seq数据的系统列表不断增长,用户可能有兴趣对每个空间体素voxels进行“反卷积”以预测细胞类型的潜在组成。在准备此小插图时,我们使用了参考scRNA-seq数据集测试了多种反卷积和整合方法decovonlution and integration
反褶积是对一个信号进行滤波以补偿一个不需要的卷积的过程。反褶积的目的是重现卷积之前的信号。
使用SMART-Seq2协议生成的来自Allen研究所的约14,000个成年小鼠皮质细胞分类学。我们一直发现使用整合方法integration (而不是反卷积方法)具有优越的性能,这可能是由于描述 空间和单细胞数据集的特征的 噪声模型存在很大差异,并且整合方法经过专门设计以对这些差异具有稳健性。
因此,我们应用了Seurat v3中最近引入的基于“锚”的集成工作流 ‘anchor’-based integration workflow ,该工作流使注释能够从reference到查询集的概率转移probabilistic transfer。因此,我们利用sctransform归一化方法遵循此处介绍的标签转移工作流程,但期望新的方法来将被开发完成此任务。
我们首先加载数据(可在此处下载),对scRNA-seq参考进行预处理,然后执行标签转移。该过程为每个点输出每个scRNA-seq派生类的概率分类(a probabilistic classification for each of the scRNA-seq derived classes)。我们将这些预测添加作为Seurat对象中的新测定。
allen_reference <- readRDS("~/Downloads/allen_cortex.rds")
# note that setting ncells=3000 normalizes the full dataset but learns noise models on 3k cells
# this speeds up SCTransform dramatically with no loss in performance
library(dplyr)
allen_reference <- SCTransform(allen_reference, ncells = 3000, verbose = FALSE) %>% RunPCA(verbose = FALSE) %>%
RunUMAP(dims = 1:30)
# After subsetting, we renormalize cortex
cortex <- SCTransform(cortex, assay = "Spatial", verbose = FALSE) %>% RunPCA(verbose = FALSE)
# the annotation is stored in the 'subclass' column of object metadata
DimPlot(allen_reference, group.by = "subclass", label = TRUE)
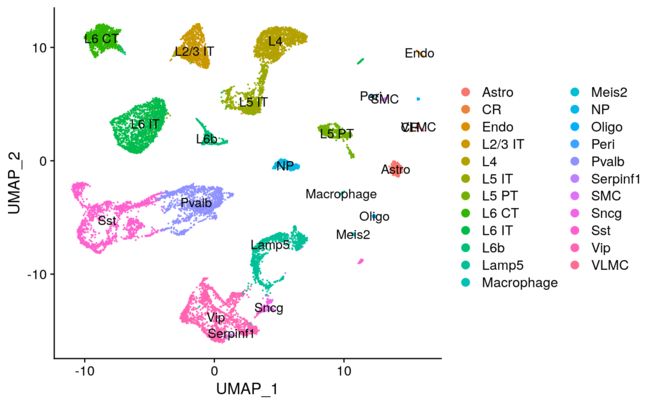
嗯,然后是经典的FindAnchor
anchors <- FindTransferAnchors(reference = allen_reference, query = cortex, normalization.method = "SCT")
predictions.assay <- TransferData(anchorset = anchors, refdata = allen_reference$subclass, prediction.assay = TRUE,
weight.reduction = cortex[["pca"]])
cortex[["predictions"]] <- predictions.assay
现在我们获得每个class的每个的预测得分。在额叶皮层区域中特别感兴趣的是层状兴奋性神经元。在这里,我们可以区分这些神经元亚型的不同顺序层,例如:的预测得分。在额叶皮层区域中特别感兴趣的是层状兴奋性神经元。在这里,我们可以区分这些神经元亚型的不同顺序层sequential layers ,例如:
DefaultAssay(cortex) <- "predictions"
SpatialFeaturePlot(cortex, features = c("L2/3 IT", "L4"), pt.size.factor = 1.6, ncol = 2, crop = TRUE)
基于这些预测分数,我们还可以预测位置受空间限制的细胞类型。我们使用基于marked point processes的相同方法来定义空间可变特征,但是是将细胞类型预测得分用作“marks”而不是基因表达。
cortex <- FindSpatiallyVariableFeatures(cortex, assay = "predictions", features = rownames(cortex),
r.metric = 5, slot = "data")
top.clusters <- head(SpatiallyVariableFeatures(cortex), 4)
SpatialPlot(object = cortex, features = top.clusters, ncol = 2)
最后,我们证明我们的整合程序能够恢复神经元和非神经元子集的已知空间定位模式,包括层状兴奋性、 layer-1 星形胶质细胞和皮质灰质。
十一、在Seurat中处理多个切片
小鼠大脑的该数据集包含与大脑另一半相对应的另一个切片。在这里,我们将其读入并执行相同的初始归一化。
brain2 <- LoadData("stxBrain", type = "posterior1")
brain2 <- SCTransform(brain2, assay = "Spatial", verbose = FALSE)
为了在同一个Seurat对象中使用多个切片,我们提供了该merge函数。
brain.merge <- merge(brain, brain2)
然后,这就使得能够用联合降维并在RNA表达数据的基础上聚类。
DefaultAssay(brain.merge) <- "SCT"
VariableFeatures(brain.merge) <- c(VariableFeatures(brain), VariableFeatures(brain2))
brain.merge <- RunPCA(brain.merge, verbose = FALSE)
brain.merge <- FindNeighbors(brain.merge, dims = 1:30)
brain.merge <- FindClusters(brain.merge, verbose = FALSE)
brain.merge <- RunUMAP(brain.merge, dims = 1:30)
最后,可以在单个UMAP图中共同进行数据可视化。SpatialDimPlot和SpatialFeaturePlot默认情况下会将所有切片绘制为列,将groupings/features绘制为行。
DimPlot(brain.merge, reduction = "umap", group.by = c("ident", "orig.ident"))

SpatialDimPlot(brain.merge)
SpatialFeaturePlot(brain.merge, features = c("Hpca", "Plp1"))
参考文章
- Seurat v3.1.2官网
- Seurat 新版教程:分析空间转录组数据