在使用ggplot2进行绘图之前,我们需要先把数据整理成规范的格式。在R语言入门中我们了解到,ggplot2只支持tidy格式的数据,因此我们需要先转换数据格式。
1.设置工作目录
# 使用setwd命令设置工作目录
setwd("H:/GeneK/ggplot")
# 使用getwd命令查看工作目录是否设置成功
getwd()
注意:在Windows上设置工作路径时,记得要把从Windows资源管理器中复制过来的路径中的符号 \ 改成 / ,否则Rstudio无法识别,会报错。如:
我从Windows中直接复制过来的地址是:H:\GeneK\ggplot,必须修改成H:/GeneK/ggplotRstudio才能识别。
2.读取数据
使用R的read.table读取相关数据:
# 读取基因表达信息
gene_exp <- read.table(file = "gene_exp.txt",
sep = "\t",
header = T,
stringsAsFactors = F)
# 读取基因长度注释信息
gene_len <- read.table(file = "gene_len.txt",
sep = "\t",
header = T,
stringsAsFactors = F)
# 读取样品分组信息
group <- read.table(file = "group.txt",
sep = "\t",
header = T,
stringsAsFactors = F)
读取结果如下:
1.基因表达信息

基因表达信息
2.基因长度注释信息
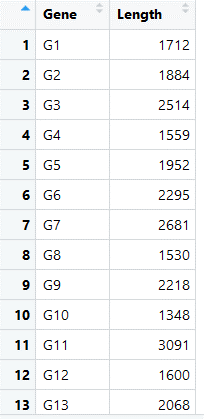
基因长度注释信息
3.样品分组信息
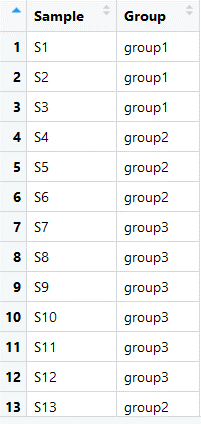
样品分组信息
3.数据变形
(1)将数据转换成Tidy data格式
由基因的表达信息表格我们可以看到,该表格不符合tidy data的原则,需要使用Tidyr包对数据格式进行调整。
# 加载Tidyr包
library(tidyr)
#使用gather函数将数据整理为Tidy格式
dexp1 <- gather(gene_exp,
key = Sample,
value = Expression,
-Gene)
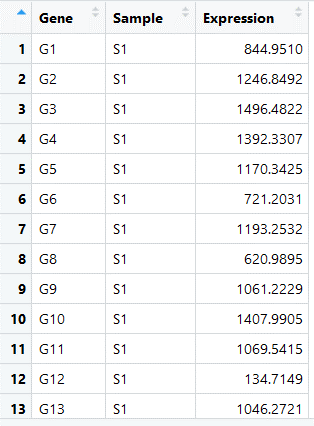
image
(2)添加基因注释和样品分组信息
#加载dplyr包
library(dplyr)
#使用left_jion添加基因长度注释信息
dexp2 <- left_join(dexp1, gene_len, by = "Gene")
#使用left_jion添加样品分组信息
dexp3 <- left_join(dexp2, group, by = "Sample")
#使表格的列按Gene,Sample,Group,Expression,Length的顺序排列
dexp4 <- select(dexp3, Gene, Sample, Group, Expression, Length)
#使表格按照基因名升序排列
dexp <- arrange(dexp4, Gene)
最终的呈现结果:

image
当然,我们还可以使用管道操作符,把上面的各步操作连接起来,一步完成转换:
dexp <- gather(gene_exp,
key = Sample,
value = Expression,
-Gene)%>%
left_join(gene_len, by = "Gene")%>%
left_join(group, by = "Sample") %>%
select(Gene, Sample, Group, Expression, Length)%>%
arrange(Gene)
注:本文是我在基因课网络课程《ggplot:绘图的艺术》的学习笔记,使用Markdown写作。