原视频6:测序质量的控制
首先建立文件夹
$ cd ~/project/wes/
$ mkdir {raw,clean,align,mutation,qc}
这部分包括fastqc和multiqc两个软件查看测序质量,以及使用trim_galore软件进行过滤低质量reads和去除接头。
1 QC
1.1 fastqc
没有原视频中文件,我用了下载的三个文件做例子。乳腺癌的组织样本。所以原视频中命令我也用不上,但是还是列出来
find /public/project/wes/raw_data -name *.gz|grep -v '\._'|xargs fastqc -t 10 -o ./
想知道为什么要-v ._,去看原视频中的文件命名。
我的文件没那么复杂,可以下面这样
$ find *.gz|xargs fastqc -t 20
-t 20 一次运行20个文件。
如果你有很多很多文件,参考我这篇批量对多个测序文件进行fastqc.
1.2 multiqc
$ multiqc -n wes ./
......
[INFO ] multiqc : This is MultiQC v1.0.dev0
[INFO ] multiqc : Template : default
[INFO ] multiqc : Searching './'
[INFO ] fastqc : Found 8 reports
[INFO ] multiqc : Report : wes.html
[INFO ] multiqc : Data : wes_data
[INFO ] multiqc : MultiQC complete
结果如下所示:
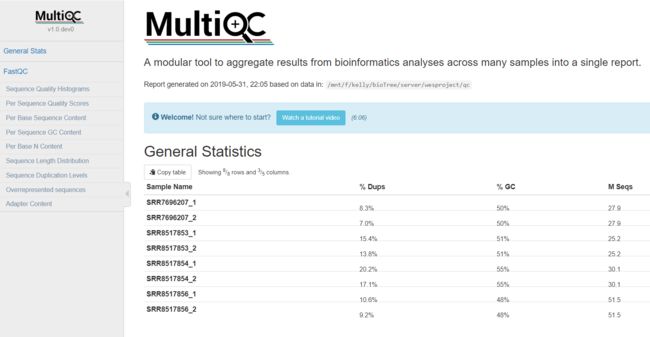
multiqc结果
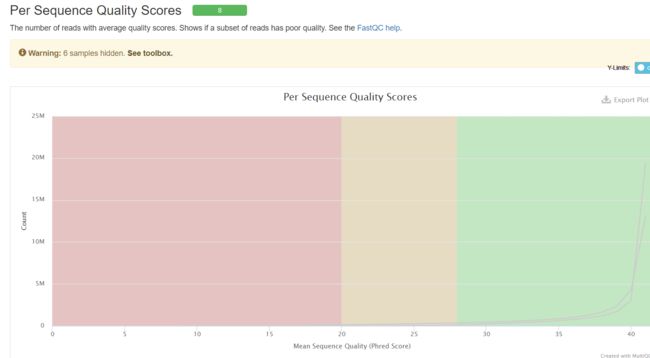
Per Sequence Quality Scores
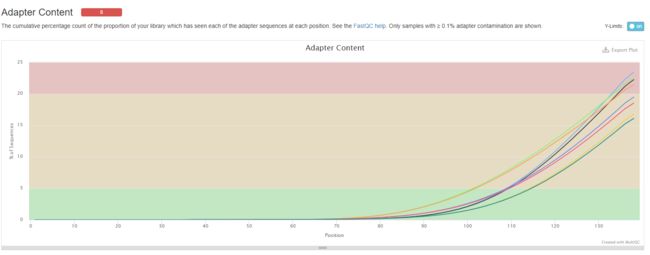
接头
以上结果发现,质量可以但是需要去接头
2 trim-galore 过滤低质量reads和去接头
Trim Galore! is a wrapper script to automate quality and adapter trimming as well as quality control
#安装
conda install -c bioconda trim-galore
ls /path/to/your/directory/*_1.fastq.gz >1
ls /path/to/your/directory/*_2.fastq.gz >2
paste 1 2 > config
也可以用
ls|grep >
打开qc.sh,写入以下内容
source activate wes
bin_trim_galore=trim_galore
dir='/mnt/f/kelly/bioTree/server/wesproject/raw_data'
cat config |while read id
do
arr=(${id})
fq1=${arr[0]}
fq2=${arr[1]}
nohup $bin_trim_galore -q 25 --phred33 --length 36 -e 0.1 --stringency 3 --paired -o $dir $fq1 $fq2 &
done
运行上面脚本bash qc.sh就可以了。
可以看到后台正在运行
kelly@DESKTOP-MRA1M1F:~$ ps -ef
UID PID PPID C STIME TTY TIME CMD
root 1 0 0 17:06 ? 00:00:00 /init
kelly 2 1 0 17:06 tty1 00:00:01 -bash
kelly 527 1 5 22:46 tty1 00:00:27 perl /home/kelly/miniconda3/bin/trim_galore -q 25 --phred33 --length 36
kelly 528 1 5 22:46 tty1 00:00:30 perl /home/kelly/miniconda3/bin/trim_galore -q 25 --phred33 --length 36
kelly 529 1 5 22:46 tty1 00:00:29 perl /home/kelly/miniconda3/bin/trim_galore -q 25 --phred33 --length 36
kelly 530 1 5 22:46 tty1 00:00:26 perl /home/kelly/miniconda3/bin/trim_galore -q 25 --phred33 --length 36
运行时间大概4h,生成以下几个文件
├── [ 88] 1
├── [ 88] 2
├── [ 176] config
├── [ 50K] nohup.out
├── [2.2G] SRR7696207_1.fastq.gz
├── [4.8K] SRR7696207_1.fastq.gz_trimming_report.txt
├── [2.1G] SRR7696207_1_val_1.fq.gz
├── [2.4G] SRR7696207_2.fastq.gz
├── [5.0K] SRR7696207_2.fastq.gz_trimming_report.txt
├── [2.3G] SRR7696207_2_val_2.fq.gz
├── [1.9G] SRR8517853_1.fastq.gz
├── [4.8K] SRR8517853_1.fastq.gz_trimming_report.txt
├── [1.8G] SRR8517853_1_val_1.fq.gz
├── [2.1G] SRR8517853_2.fastq.gz
├── [5.0K] SRR8517853_2.fastq.gz_trimming_report.txt
├── [2.0G] SRR8517853_2_val_2.fq.gz
├── [2.3G] SRR8517854_1.fastq.gz
├── [4.8K] SRR8517854_1.fastq.gz_trimming_report.txt
├── [2.2G] SRR8517854_1_val_1.fq.gz
├── [2.6G] SRR8517854_2.fastq.gz
├── [5.1K] SRR8517854_2.fastq.gz_trimming_report.txt
├── [2.4G] SRR8517854_2_val_2.fq.gz
├── [4.1G] SRR8517856_1.fastq.gz
├── [4.9K] SRR8517856_1.fastq.gz_trimming_report.txt
├── [4.0G] SRR8517856_1_val_1.fq.gz
├── [4.5G] SRR8517856_2.fastq.gz
├── [5.1K] SRR8517856_2.fastq.gz_trimming_report.txt
├── [4.2G] SRR8517856_2_val_2.fq.gz
└── [ 305] trim
可见,各小了大概0.1G。
其中,txt中的信息如下
SUMMARISING RUN PARAMETERS
==========================
Input filename: SRR7696207_1.fastq.gz
Trimming mode: paired-end
Trim Galore version: 0.6.2
Cutadapt version: 1.18
Number of cores used for trimming: 1
Quality Phred score cutoff: 25
Quality encoding type selected: ASCII+33
Adapter sequence: 'AGATCGGAAGAGC' (Illumina TruSeq, Sanger iPCR; auto-detected)
Maximum trimming error rate: 0.1 (default)
Minimum required adapter overlap (stringency): 3 bp
Minimum required sequence length for both reads before a sequence pair gets removed: 36 bp
Output file will be GZIP compressed
This is cutadapt 1.18 with Python 2.7.15
Command line parameters: -j 1 -e 0.1 -q 25 -O 3 -a AGATCGGAAGAGC SRR7696207_1.fastq.gz
Processing reads on 1 core in single-end mode ...
Finished in 1011.58 s (36 us/read; 1.65 M reads/minute).
=== Summary ===
Total reads processed: 27,850,979
Reads with adapters: 9,452,510 (33.9%)
Reads written (passing filters): 27,850,979 (100.0%)
Total basepairs processed: 4,177,646,850 bp
Quality-trimmed: 21,999,885 bp (0.5%)
Total written (filtered): 3,930,084,164 bp (94.1%)
=== Adapter 1 ===
Sequence: AGATCGGAAGAGC; Type: regular 3'; Length: 13; Trimmed: 9452510 times.
No. of allowed errors:
0-9 bp: 0; 10-13 bp: 1
Bases preceding removed adapters:
A: 21.7%
C: 25.2%
G: 28.1%
T: 24.9%
none/other: 0.0%
Overview of removed sequences
length count expect max.err error counts
3 619264 435171.5 0 619264
4 291812 108792.9 0 291812
5 236788 27198.2 0 236788
6 226036 6799.6 0 226036
7 217969 1699.9 0 217969
8 214073 425.0 0 214073
9 230269 106.2 0 229591 678
10 216909 26.6 1 209213 7696
11 223835 6.6 1 214662 9173
12 219837 1.7 1 210062 9775
13 219106 0.4 1 209421 9685
14 223045 0.4 1 212324 10721
15 218505 0.4 1 208196 10309
16 224812 0.4 1 213584 11228
17 228422 0.4 1 216425 11997
18 214056 0.4 1 204368 9688
19 216385 0.4 1 206368 10017
20 207262 0.4 1 198505 8757
21 220284 0.4 1 209515 10769
22 207937 0.4 1 198723 9214
23 203136 0.4 1 194604 8532
24 208015 0.4 1 198522 9493
25 200567 0.4 1 191904 8663
26 201338 0.4 1 192675 8663
......
后面如果想快速运行流程,可以把测序数据取前N行,那么请看
3_0_4 要理解并会用的几个脚本
如果就想运行所有数据,请到
4 比对到参考基因组输出bam文件