刘小泽写于2020.7.19
为何取名叫“交响乐”?因为单细胞分析就像一个大乐团,需要各个流程的协同配合
单细胞交响乐1-常用的数据结构SingleCellExperiment
单细胞交响乐2-scRNAseq从实验到下游简介
单细胞交响乐3-细胞质控
单细胞交响乐4-归一化
单细胞交响乐5-挑选高变化基因
单细胞交响乐6-降维
单细胞交响乐7-聚类分群
单细胞交响乐8-marker基因检测
单细胞交响乐9-细胞类型注释
单细胞交响乐9-细胞类型注释
单细胞交响乐10-数据集整合后的批次矫正
单细胞交响乐11-多样本间差异分析
单细胞交响乐12-检测Doublet
单细胞交响乐13-细胞周期推断
单细胞交响乐14-细胞轨迹推断
单细胞交响乐15-scRNA与蛋白丰度信息结合
单细胞交响乐16-处理大型数据
单细胞交响乐17-不同单细胞R包的数据格式相互转换
单细胞交响乐18-实战一 Smart-seq2
1 前言
前面的种种都是作为知识储备,但是不实战还是记不住前面的知识
这是第二个实战练习
使用了一个存在异质性的数据集,是研究小鼠大脑的 (Zeisel et al. 2015)
其中大约包含3000个细胞,包括少突胶质细胞,小胶质细胞和神经元等,使用的细胞分离平台是Fluidigm C1微流控系统,属于比较早期的系统【单细胞测序的知识】
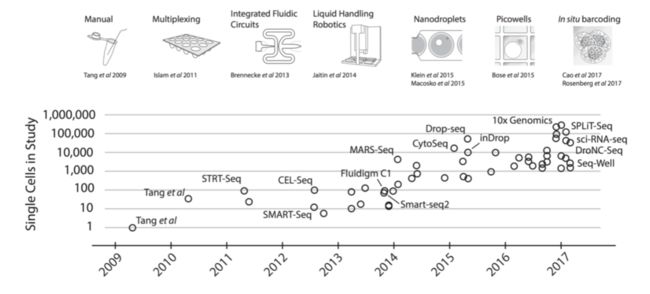
文库制备时加入了UMI
UMI简单解释:
UMI就是为了去除PCR扩增偏差的。一般一个基因对应多个UMI时,出现多个reads含有同一个UMI时,这里只计数一次。UMI英文解释:
Each transcript molecule can only produce one UMI count but can yield many reads after fragmentationUMI详细解释:
不管是bulk RNA还是scRNA,都需要进行PCR扩增,但是不可避免有一些转录本会被扩增太多次,超过了真实表达量。当起始文库很小时(比如单细胞数据),就需要更多次的PCR过程,这个次数越多,引入的误差就越大。UMI就是Unique Molecular Identifier,由4-10个随机核苷酸组成,在mRNA反转录后,进入到文库中,每一个mRNA随机连上一个UMI,根据PCR结果可以计数不同的UMI,最终统计mRNA的数量。UMI图片解释:

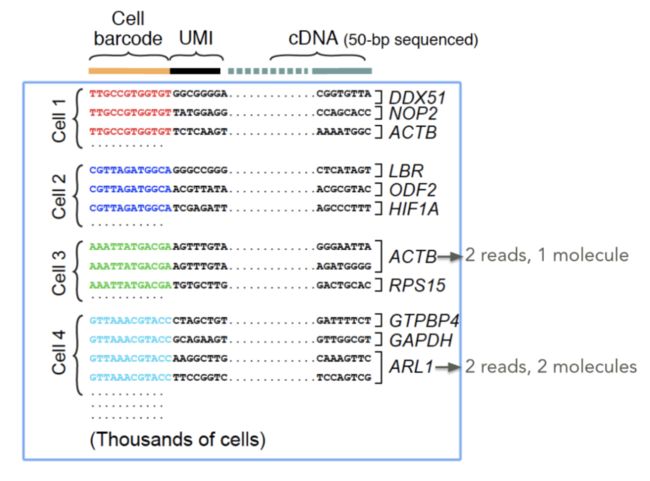
UMI有几个要求:
- 不能是均聚物 ,如AAAAAAAAAA
- 不能有N碱基
- 不能包含碱基质量低于10的碱基
2 数据准备
# 自己下载
library(scRNAseq)
sce.zeisel <- ZeiselBrainData()
# 或者使用之前分享的RData
load('sce.zeisel.RData')
sce.zeisel
# class: SingleCellExperiment
# dim: 20006 3005
# metadata(0):
# assays(1): counts
# rownames(20006): Tspan12 Tshz1 ... mt-Rnr1
# mt-Nd4l
# rowData names(1): featureType
# colnames(3005): 1772071015_C02 1772071017_G12
# ... 1772066098_A12 1772058148_F03
# colData names(10): tissue group # ...
# level1class level2class
# reducedDimNames(0):
# altExpNames(2): ERCC repeat
看到这么几个信息:2万多基因,3005个样本;只有原始count矩阵;使用了symbol ID;加入了ERCC
# 有57个ERCC
> dim(altExp(sce.zeisel,'ERCC'))
[1] 57 3005
# 而且已经标注了线粒体基因
> table(rowData(sce.zeisel))
endogenous mito
19972 34
一个重要操作:aggregateAcrossFeatures
英文解释是:Sum together expression values (by default, counts) for each feature set in each cell.
但是只看说明还是不好理解,举个例子:
可以看到下面会有很多基因具有多个loc
比如OTTMUSG00000016609_loc4、OTTMUSG00000016609_loc3 其实可以算作一个基因
head(rownames(sce.zeisel)[grep("_loc[0-9]+$",rownames(sce.zeisel))])
# [1] "Syne1_loc2" "Hist1h2ap_loc1"
# [3] "Inadl_loc1" "OTTMUSG00000016609_loc4"
# [5] "OTTMUSG00000016609_loc3" "Gm5643_loc2"
# 这样的有300多个
> length(grep("_loc[0-9]+$",rownames(sce.zeisel)))
[1] 330
如果拿一个基因来看
Syne1有Syne1_loc1和Syne1_loc2
> length(grep("Syne1",rownames(sce.zeisel)))
[1] 2
counts(sce.zeisel)[grep("Syne1",rownames(sce.zeisel)),][1:2,1:3]
# 1772071015_C02 1772071017_G12 1772071017_A05
# Syne1_loc2 11 2 4
# Syne1_loc1 0 0 4
如果使用这个函数,会有怎样效果
test <- aggregateAcrossFeatures(sce.zeisel,
id=sub("_loc[0-9]+$", "", rownames(sce.zeisel)))
# 只剩一个了,也就是合二为一
> length(grep("Syne1",rownames(test)))
[1] 1
# 看表达量,也是合二为一
> counts(test)[grep("Syne1",rownames(test)),][1:3]
1772071015_C02 1772071017_G12 1772071017_A05
11 2 8
因此,明白了,这个函数就是处理相同行:把几个相同的行的值加在一起变为一行
也就明白了,下面为什么要进行sub操作,其实就是为了把loc去掉,暴露出相同的基因名,才能执行aggregateAcrossFeatures函数
sce.zeisel <- aggregateAcrossFeatures(sce.zeisel,
id=sub("_loc[0-9]+$", "", rownames(sce.zeisel)))
> dim(sce.zeisel)
[1] 19839 3005
再添加Ensembl ID
library(org.Mm.eg.db)
rowData(sce.zeisel)$Ensembl <- mapIds(org.Mm.eg.db,
keys=rownames(sce.zeisel), keytype="SYMBOL", column="ENSEMBL")
3 质控
还是备份一下,把unfiltered数据主要用在质控的探索上
unfiltered <- sce.zeisel
这个公共数据的作者在发表文章时将数据的低质量细胞去掉了,但并不妨碍我们做个质控,也可以看看它去除的怎样
stats <- perCellQCMetrics(sce.zeisel, subsets=list(
Mt=rowData(sce.zeisel)$featureType=="mito"))
qc <- quickPerCellQC(stats, percent_subsets=c("altexps_ERCC_percent",
"subsets_Mt_percent"))
sce.zeisel <- sce.zeisel[,!qc$discard]
> sum(qc$discard)
[1] 189
> dim(sce.zeisel)
[1] 19839 2816
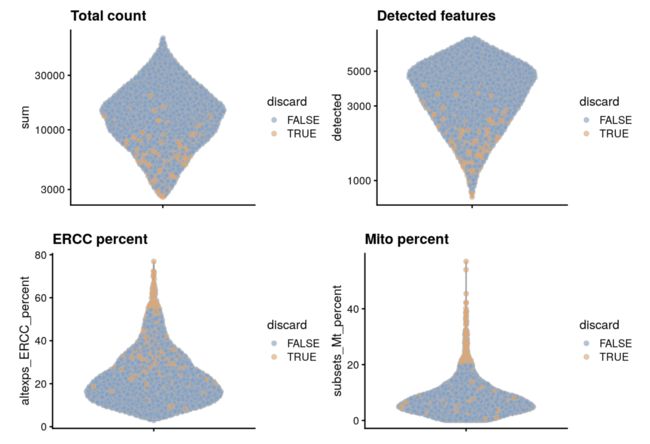
根据原来的数据,加上质控标准作图
colData(unfiltered) <- cbind(colData(unfiltered), stats)
unfiltered$discard <- qc$discard
# 做个图
gridExtra::grid.arrange(
plotColData(unfiltered, y="sum", colour_by="discard") +
scale_y_log10() + ggtitle("Total count"),
plotColData(unfiltered, y="detected", colour_by="discard") +
scale_y_log10() + ggtitle("Detected features"),
plotColData(unfiltered, y="altexps_ERCC_percent",
colour_by="discard") + ggtitle("ERCC percent"),
plotColData(unfiltered, y="subsets_Mt_percent",
colour_by="discard") + ggtitle("Mito percent"),
ncol=2
)
再看下文库大小和ERCC分别和线粒体含量的关系
gridExtra::grid.arrange(
plotColData(unfiltered, x="sum", y="subsets_Mt_percent",
colour_by="discard") + scale_x_log10(),
plotColData(unfiltered, x="altexps_ERCC_percent", y="subsets_Mt_percent",
colour_by="discard"),
ncol=2
)
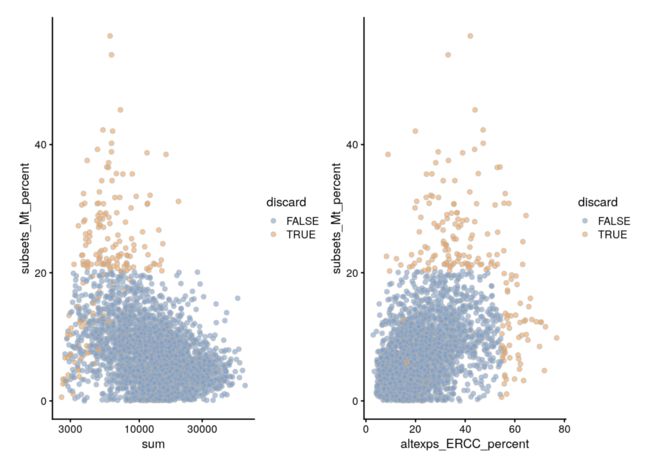
然后检查一下被过滤的原因
## low_lib_size low_n_features high_altexps_ERCC_percent
## 0 3 65
## high_subsets_Mt_percent discard
## 128 189
4 归一化
这里细胞数量较多,因此需要预先分群+去卷积计算size factor
library(scran)
set.seed(1000)
clusters <- quickCluster(sce.zeisel)
sce.zeisel <- computeSumFactors(sce.zeisel, cluster=clusters)
sce.zeisel <- logNormCounts(sce.zeisel)
summary(sizeFactors(sce.zeisel))
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.119 0.486 0.831 1.000 1.321 4.509
看看两种归一化方法的差异
# 常规:最简单的只考虑文库大小
summary(librarySizeFactors(sce.zeisel))
# Min. 1st Qu. Median Mean 3rd Qu. Max.
# 0.1757 0.5680 0.8680 1.0000 1.2783 4.0839
plot(librarySizeFactors(sce.zeisel), sizeFactors(sce.zeisel), pch=16,
xlab="Library size factors", ylab="Deconvolution factors", log="xy")

5 找高变异基因
理论上,应该对每个细胞都标记批次信息,添加block信息。但是这里由于技术不同,每个板子上只有20-40个细胞,并且细胞群体具有高度异质性,不能假设每个板上的细胞类型的分布是相同的,因此这里不使用block将批次信息“锁住”是合适的
既然有ERCC,就可以用第三种方法【在之前单细胞交响乐5-挑选高变化基因的2.3 考虑技术噪音】:
dec.zeisel <- modelGeneVarWithSpikes(sce.zeisel, "ERCC")
top.hvgs <- getTopHVGs(dec.zeisel, prop=0.1)
> length(top.hvgs)
[1] 1816
同样使用了spike-in,对比一下,看到这里不管总体方差还是技术因素方差都要比之前smart-seq2要小。
smart-seq2是按read计数,这里由于添加了UMI,是按molecule计数,也就是说,UMI的加入,确实减少了PCR扩增的偏差影响
另外图中看到,这里STRT-seq的spike-in方差一直要比内源基因的方差小,也就是说内源基因的变化幅度一直保持高位,体现了数据中包含多种细胞类型而导致的异质性,异质性导致了基因表达极度不均衡

6 降维
library(BiocSingular)
set.seed(101011001)
sce.zeisel <- denoisePCA(sce.zeisel, technical=dec.zeisel, subset.row=top.hvgs)
sce.zeisel <- runTSNE(sce.zeisel, dimred="PCA")
看一下得到的PC数
ncol(reducedDim(sce.zeisel, "PCA"))
## [1] 50
7 聚类
snn.gr <- buildSNNGraph(sce.zeisel, use.dimred="PCA")
colLabels(sce.zeisel) <- factor(igraph::cluster_walktrap(snn.gr)$membership)
看一下结果
table(colLabels(sce.zeisel))
##
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14
## 283 451 114 143 599 167 191 128 350 70 199 58 39 24
画图
plotTSNE(sce.zeisel, colour_by="label")

8 找marker基因并解释结果
主要还是关注上调基因,可以帮我们快速判断出异质性群体中各个细胞类型的差异
比如还是针对cluster1看看
markers <- findMarkers(sce.zeisel, direction="up")
marker.set <- markers[["1"]]
> ncol(marker.set)
[1] 17
> colnames(marker.set)
[1] "Top" "p.value" "FDR" "summary.logFC" "logFC.2" "logFC.3" "logFC.4"
[8] "logFC.5" "logFC.6" "logFC.7" "logFC.8" "logFC.9" "logFC.10" "logFC.11"
[15] "logFC.12" "logFC.13" "logFC.14"
head(marker.set[,1:8], 10)
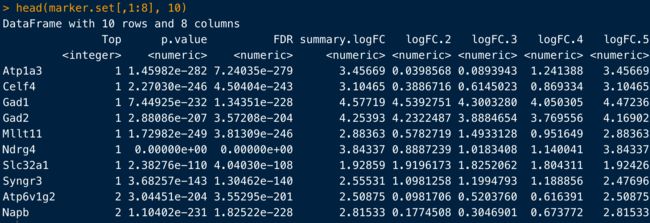
使用cluster1的Top10基因(但不一定只是10个)画热图
top.markers <- rownames(marker.set)[marker.set$Top <= 10]
> length(top.markers)
[1] 58
plotHeatmap(sce.zeisel, features=top.markers, order_columns_by="label")
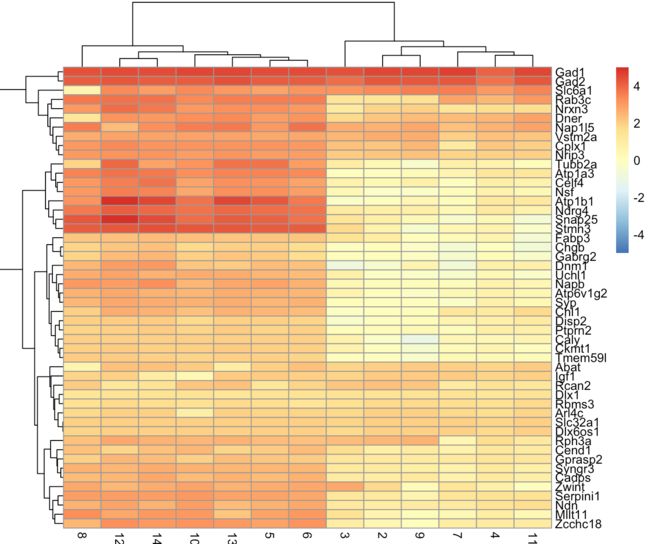
接下来就是根据背景知识了,比如看到Gad1、Slc6a1表达量都很高,可能表明cluster1属于中间神经元

另一种方法:基于logFC
比如可以挑出cluster1的计算结果marker.set中前50个基因(这里就是50个,而不是Top50),然后根据cluster1与其他clusters的logFC,对每个基因表达量做热图
library(pheatmap)
logFCs <- getMarkerEffects(marker.set[1:50,])
pheatmap(logFCs, breaks=seq(-5, 5, length.out=101))
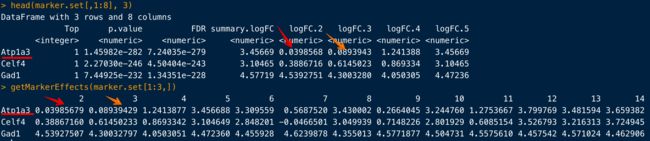
那么这个函数到底做了什么呢?看图就知道:
也就是把每个基因在其他clusters的logFC结果挑出来,汇集成了一个新矩阵,我们自己手动也是可以做到的
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
