scATAC分析神器ArchR初探-简介(1)
scATAC分析神器ArchR初探-ArchR进行doublet处理(2)
scATAC分析神器ArchR初探-创建ArchRProject(3)
scATAC分析神器ArchR初探-使用ArchR降维(4)
scATAC分析神器ArchR初探--使用ArchR进行聚类(5)
scATAC分析神器ArchR初探-单细胞嵌入(6)
scATAC分析神器ArchR初探-使用ArchR计算基因活性值和标记基因(7)
scATAC分析神器ArchR初探-scRNA-seq确定细胞类型(8)
scATAC分析神器ArchR初探-ArchR中的伪批次重复处理(9)
scATAC分析神器ArchR初探-使用ArchR-peak-calling(10)
scATAC分析神器ArchR初探-使用ArchR识别标记峰(11)
scATAC分析神器ArchR初探-使用ArchR进行主题和功能丰富(12)
scATAC分析神器ArchR初探-利用ArchR丰富ChromVAR偏差(13)
scATAC分析神器ArchR初探-使用ArchR进行足迹(14)
scATAC分析神器ArchR初探-使用ArchR进行整合分析(15)
scATAC分析神器ArchR初探-使用ArchR进行轨迹分析(16)
7使用ArchR进行基因评分和标记基因
尽管ArchR能够可靠地调用集群,但无法事先知道每个集群代表哪种小区类型。由于每个应用程序都是不同的,因此通常将这项任务留给手动注释。
为了进行这种细胞类型注释,我们使用了细胞类型特异性标记基因的先验知识,并且我们通过使用基因评分从染色质可及性数据中估计了这些基因的基因表达。基因得分本质上是基于基因附近调控元件的可及性来预测基因将高度表达的预测。为了创建这些基因评分,ArchR允许使用用户提供的复杂的自定义距离加权可访问性模型。
7.1在ArchR中计算基因得分
在我们的出版物中,我们测试了50多种不同的基因评分模型,并确定了一类模型,这些模型在各种测试条件下始终优于其他模型。在ArchR中作为默认模型实现的此类模型具有三个主要组成部分:
整个基因体内的可及性有助于基因得分。
指数加权函数,以距离依赖的方式说明假定的远端调节元件的活动。
施加的基因边界可最大程度地减少无关的调控元件对基因得分的影响。
那么ArchR如何计算基因得分呢?对于每条染色体,ArchR使用未预先计算的用户定义的图块大小(默认值为500 bp)创建图块矩阵,并将这些图块与用户定义的基因窗口重叠(基因两侧的默认值为100 kb) ),然后计算从每个图块(开始或结束)到基因体(具有可选的扩展名上游或下游)或基因起始点的距离。我们已经发现,基因表达的最佳预测因子是包括启动子和基因体的基因区域的局部可及性。如上所述,为正确考虑给定基因的远端可及性,ArchR标识了位于基因窗口内且未穿过另一个基因区域的图块的子集。这种过滤允许包含远端调控元件,其可以提高预测基因表达值的准确性,但排除更可能与另一个基因(例如,附近基因的启动子)相关的调控元件。然后使用用户定义的可访问性模型(默认值为e(-abs(distance)/ 5000)+ e-1)将每个图块到基因的距离转换为距离权重。当基因体被包含在基因区域中时(基于距离的权重是最大可能的权重),我们发现非常大的基因会偏向整体基因得分。在这些情况下,由于内含子和外显子都包含插入片段,因此总基因得分可能会有很大差异。为了帮助调整基因大小的这些巨大差异,ArchR对基因大小的倒数(1 /基因大小)施加单独的权重,并将此反权重从1线性缩放到用户定义的硬最大值(默认为5)。因此,较小的基因获得较大的相对权重,部分地使这种长度效应标准化。然后,将相应的距离和基因大小权重乘以每个图块内Tn5插入的数量,并在基因窗口内的所有图块上求和,同时仍按上述方法考虑附近的基因区域。这种可访问性总和是“基因得分”,并且将所有基因的深度标准化为用户定义的常数(默认值为10,000)。然后将计算出的基因评分存储在相应的Arrow文件中,以进行下游分析。因此,较小的基因获得较大的相对权重,部分地使这种长度效应标准化。然后,将相应的距离和基因大小权重乘以每个图块内Tn5插入的数量,并在基因窗口内的所有图块上求和,同时仍按上述方法考虑附近的基因区域。这种可访问性总和是“基因得分”,并且将所有基因的深度标准化为用户定义的常数(默认值为10,000)。然后将计算出的基因评分存储在相应的Arrow文件中,以进行下游分析。因此,较小的基因获得较大的相对权重,部分地使这种长度效应标准化。然后,将相应的距离和基因大小权重乘以每个图块内Tn5插入的数量,并在基因窗口内的所有图块上求和,同时仍按上述方法考虑附近的基因区域。这种可访问性总和是“基因得分”,并且将所有基因的深度标准化为用户定义的常数(默认值为10,000)。然后将计算出的基因评分存储在相应的Arrow文件中,以进行下游分析。同时仍然说明了如上所述的附近基因区域。这种可访问性总和是“基因得分”,并且将所有基因的深度标准化为用户定义的常数(默认值为10,000)。然后将计算出的基因评分存储在相应的Arrow文件中,以进行下游分析。同时仍然说明了如上所述的附近基因区域。这种可访问性总和是“基因得分”,并且将所有基因的深度标准化为用户定义的常数(默认值为10,000)。然后将计算出的基因评分存储在相应的Arrow文件中,以进行下游分析。
为了说明默认的ArchR基因得分模型是什么样子,我们提供了这个玩具示例,显示了在整个基因区域中应用的权重:
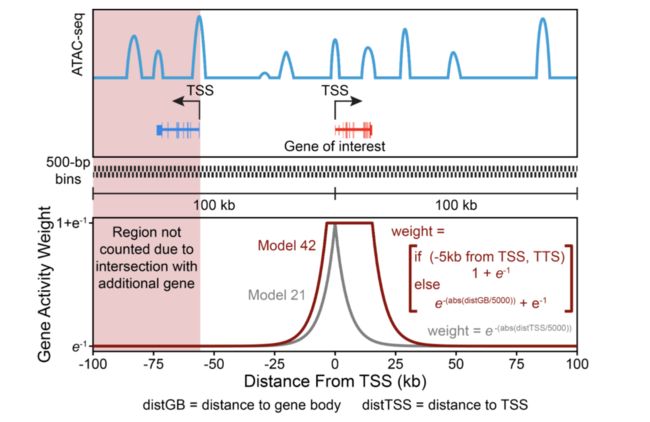
如果将参数addGeneScoreMat设置为,则在创建时会为每个Arrow文件计算基因得分TRUE-这是默认行为。或者,可以使用该addGeneScoreMatrix()功能随时将基因评分添加到Arrow文件中。一旦计算,嵌入的单个细胞可以通过它们的基因得分着色,以帮助鉴定各种细胞类型。我们将在本章的其余部分中说明基因评分的应用。
重要的是要注意,并不是所有的基因在基因评分上都表现良好。特别是,居住在基因密集区的基因可能会出现问题。因此,最好总是通过查看测序轨迹来理清所有基因得分分析,这将在下一章中进行介绍。
7.2标记功能的识别
除了使用相关标记基因的先验知识来注释簇外,ArchR还可以针对任何给定的细胞分组(例如簇)无偏地识别标记特征。这些特征可以是任何东西-峰,基因(基于基因得分)或转录因子基序(基于chromVAR偏差)。ArchR使用getMarkerFeatures()可以通过useMatrix参数将任何矩阵作为输入的函数来执行此操作,并且可以识别groupBy参数所指示的组所独有的特征。如果useMatrix参数设置为“ GeneScoreMatrix”,则该函数将识别在每种细胞类型中似乎唯一活跃的基因。这提供了一种无偏见的方式来查看预测每个集群中哪些基因处于活跃状态,并有助于集群注释。
如上所述,getMarkerFeatures()可以将相同的功能与Arrow文件中存储的任何矩阵一起使用,以标识特定于某些单元格组的功能。这是通过useMatrix参数完成的。例如,useMatrix = "TileMatrix"将鉴定对特定细胞组高度特异性的基因组区域,并useMatrix = "PeakMatrix"鉴定对特定细胞组高度特异性的峰。getMarkerFeatures()后面的章节中提供了有关如何在其他要素类型上使用该功能的示例。
7.2.1标记特征识别如何发生?
标记特征识别的此过程取决于为每个单元组选择一组偏置匹配的背景单元。在所有特征中,将每个细胞组与其自身的背景细胞组进行比较,以确定给定的细胞组是否具有明显更高的可及性。
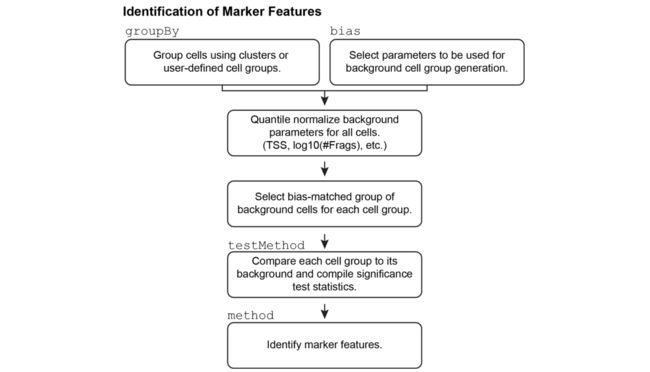
这些背景单元格组的选择对于此过程的成功至关重要,并且是在用户通过的bias参数提供的多维空间上执行的getMarkerFeatures()。对于单元组中的每个单元,ArchR会在提供的多维空间中找到不属于给定单元组成员的最近邻居单元,并将其添加到背景单元组中。这样,ArchR会创建一组与给定单元组尽可能相似的偏差匹配单元,因此即使该单元组很小,也可以更可靠地确定重要性。
ArchR这样做的方法是采用通过bias参数提供的所有尺寸,并对其值进行分位数归一化,以将每个尺寸的方差分布在相同的相对比例上。以一个玩具示例为例,如果将参数TSS和log10(Num Fragments)提供给bias,则分位数前的标准化值可能如下所示:
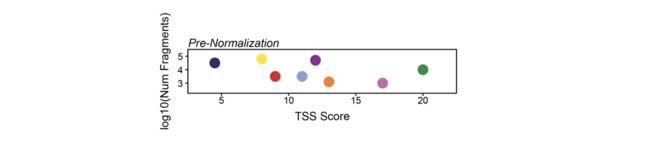
在此,与沿x轴的方差相比,沿y轴的相对方差非常小。如果我们对这些轴进行归一化处理,以使它们的值范围从0到1,则我们使相对方差更加相等。重要的是,我们还按照此图的右侧所示,极大地改变了最近的邻居。
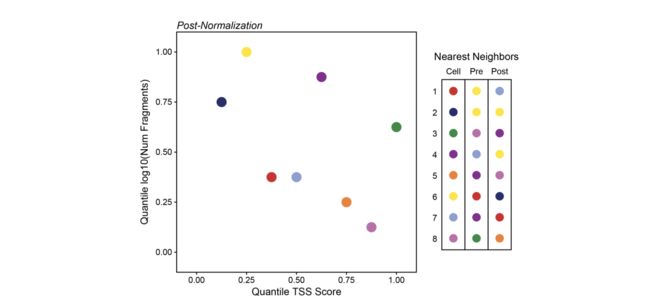
ArchR对所有维度进行归一化,并在此归一化多维空间中使用欧氏距离来找到最接近的邻居。
7.3鉴定标记基因
为了根据基因得分识别标记基因,我们用调用getMarkerFeatures()函数useMatrix = "GeneScoreMatrix"。我们指定我们想知道特定于群集的功能,groupBy = "Clusters"这些功能告诉ArchR在其中使用“群集”列cellColData对单元组进行分层。
markersGS <- getMarkerFeatures(
ArchRProj = projHeme2,
useMatrix = "GeneScoreMatrix",
groupBy = "Clusters",
bias = c("TSSEnrichment", "log10(nFrags)"),
testMethod = "wilcoxon"
)
此函数返回一个SummarizedExperiment对象,其中包含有关所标识的标记特征的相关信息。这种返回值在ArchR中很常见,并且是ArchR启用下游数据分析的关键方式之一。SummarizedExperiment对象类似于矩阵,其中行表示感兴趣的特征(即基因),而列表示样本。一个SummarizedExperiment对象包含一个或多个测定,每个测定均由数字数据的类似矩阵的对象表示,以及适用于测定矩阵行或列的元数据。深入研究SummarizedExperiment对象超出了本教程的范围,但如果需要更多信息,请查看生物导体页面。
我们可以DataFrame使用getMarkers()函数获得一个对象列表,每个集群一个,包含相关的标记功能:
markerList <- getMarkers(markersGS, cutOff = "FDR <= 0.01 & Log2FC >= 1.25")
markerList$C6
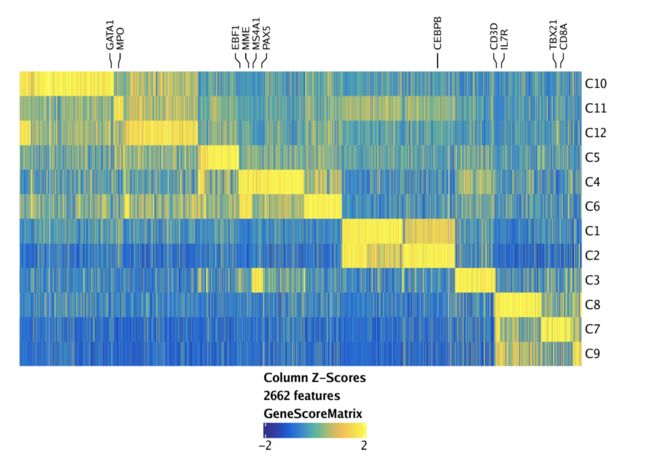
为了同时可视化所有标记特征,我们可以使用markerHeatmap()函数创建一个热图,可以选择通过labelMarkers参数提供一些标记基因在热图中进行标记。
markerGenes <- c(
"CD34", #Early Progenitor
"GATA1", #Erythroid
"PAX5", "MS4A1", "EBF1", "MME", #B-Cell Trajectory
"CD14", "CEBPB", "MPO", #Monocytes
"IRF8",
"CD3D", "CD8A", "TBX21", "IL7R" #TCells
)
heatmapGS <- markerHeatmap(
seMarker = markersGS,
cutOff = "FDR <= 0.01 & Log2FC >= 1.25",
labelMarkers = markerGenes,
transpose = TRUE
)
要绘制此热图,我们可以使用ComplexHeatmap::draw()函数,因为该heatmapGS对象实际上是热图列表:
ComplexHeatmap::draw(heatmapGS, heatmap_legend_side = "bot", annotation_legend_side = "bot")
要保存此图的可编辑矢量化版本,请使用plotPDF()。
plotPDF(heatmapGS, name = "GeneScores-Marker-Heatmap", width = 8, height = 6, ArchRProj = projHeme2, addDOC = FALSE)
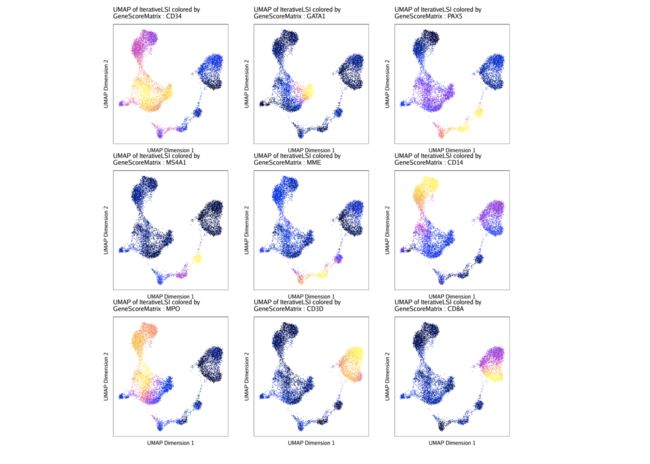
7.4可视化嵌入中的标记基因
如前所述,我们可以在UMAP嵌入中叠加每个细胞的基因得分。这是通过使用函数中的colorBy和name参数来完成的plotEmbedding()。
markerGenes <- c(
"CD34", #Early Progenitor
"GATA1", #Erythroid
"PAX5", "MS4A1", "MME", #B-Cell Trajectory
"CD14", "MPO", #Monocytes
"CD3D", "CD8A"#TCells
)
p <- plotEmbedding(
ArchRProj = projHeme2,
colorBy = "GeneScoreMatrix",
name = markerGenes,
embedding = "UMAP",
quantCut = c(0.01, 0.95),
imputeWeights = NULL
)
要绘制特定基因,我们可以将该图列表子集化:
p$CD14
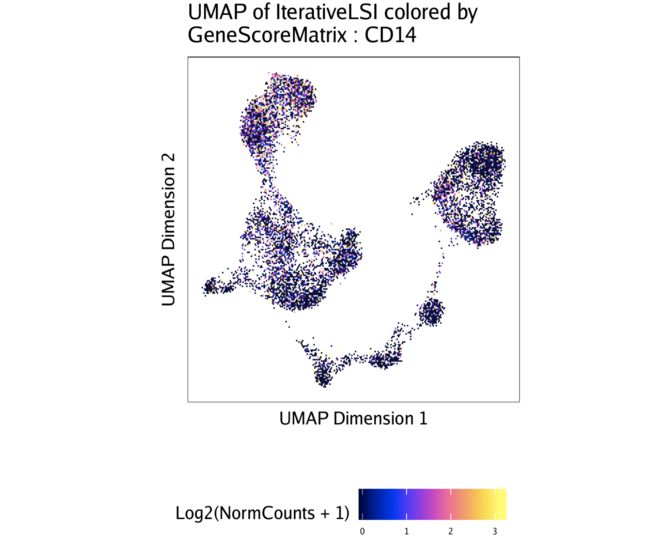
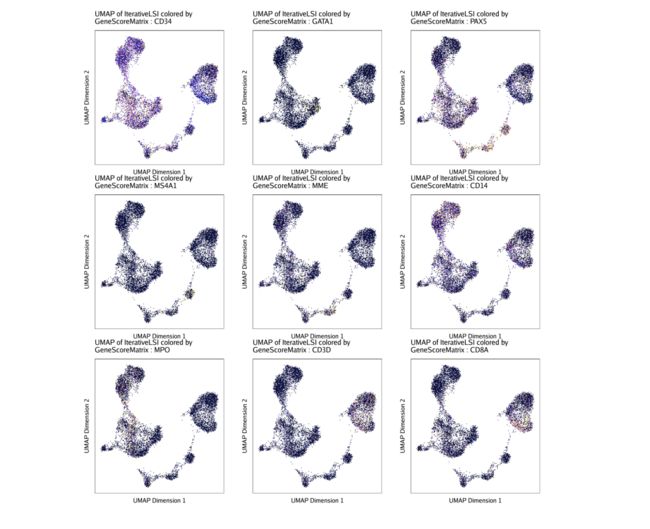
为了绘制所有基因,我们可以使用cowplot将各种标记基因排列到一个图中。
p2 <- lapply(p, function(x){
x + guides(color = FALSE, fill = FALSE) +
theme_ArchR(baseSize = 6.5) +
theme(plot.margin = unit(c(0, 0, 0, 0), "cm")) +
theme(
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)
})
do.call(cowplot::plot_grid, c(list(ncol = 3),p2))
要保存此图的可编辑矢量化版本,我们使用plotPDF()函数。
plotPDF(plotList = p,
name = "Plot-UMAP-Marker-Genes-WO-Imputation.pdf",
ArchRProj = projHeme2,
addDOC = FALSE, width = 5, height = 5)
7.5用MAGIC进行标记基因插补
在上一节中,您可能已经注意到,某些基因得分图似乎变化很大。这是因为scATAC-seq数据稀疏。我们可以使用MAGIC通过平滑附近细胞之间的信号来估算基因得分。在我们手中,这大大改善了基因评分的视觉解释。为此,我们首先将估算权重添加到ArchRProject。
projHeme2 <- addImputeWeights(projHeme2)
然后,plotEmbedding()在绘制覆盖在UMAP嵌入上的基因得分时,可以将这些估算权重传递给。
markerGenes <- c(
"CD34", #Early Progenitor
"GATA1", #Erythroid
"PAX5", "MS4A1", "MME", #B-Cell Trajectory
"CD14", "MPO", #Monocytes
"CD3D", "CD8A"#TCells
)
p <- plotEmbedding(
ArchRProj = projHeme2,
colorBy = "GeneScoreMatrix",
name = markerGenes,
embedding = "UMAP",
imputeWeights = getImputeWeights(projHeme2)
)
和以前一样,我们可以将此图列表子集来选择特定基因。
p$CD14
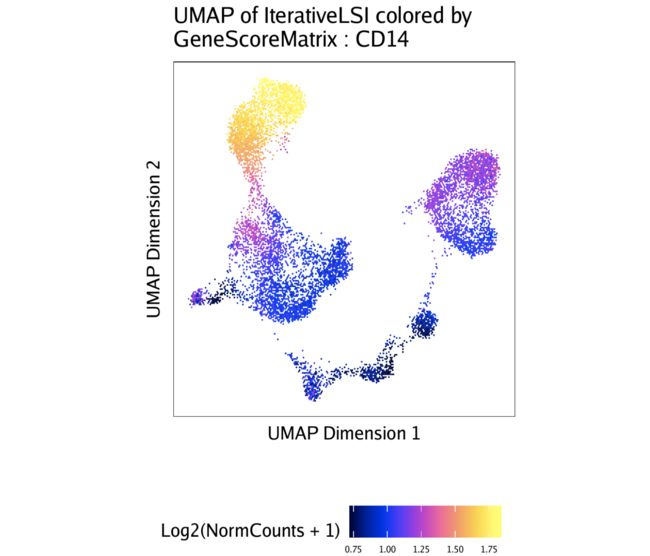
或者我们可以使用一次性绘制所有标记基因cowplot。
#Rearrange for grid plotting
p2 <- lapply(p, function(x){
x + guides(color = FALSE, fill = FALSE) +
theme_ArchR(baseSize = 6.5) +
theme(plot.margin = unit(c(0, 0, 0, 0), "cm")) +
theme(
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)
})
do.call(cowplot::plot_grid, c(list(ncol = 3),p2))
要保存此图的可编辑矢量化版本,我们使用plotPDF()函数。
plotPDF(plotList = p,
name = "Plot-UMAP-Marker-Genes-W-Imputation.pdf",
ArchRProj = projHeme2,
addDOC = FALSE, width = 5, height = 5)
7.6使用ArchRBrowser进行轨迹图
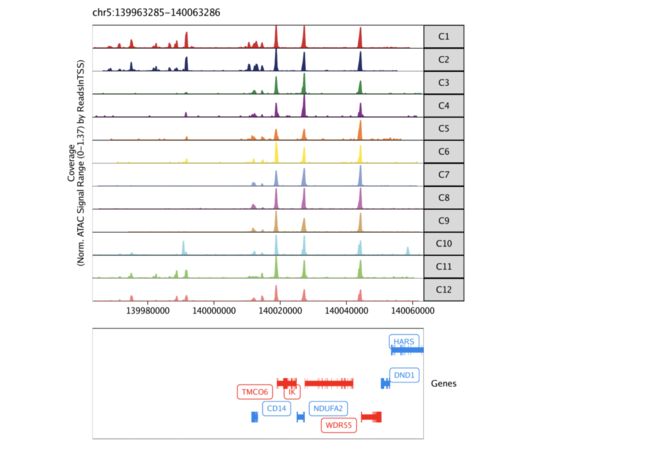
除了以UMAP覆盖图的形式绘制每个细胞的基因得分外,我们还可以使用基因组浏览器轨迹在每个簇的基础上浏览这些标记基因的局部染色质可及性。为此,我们使用plotBrowserTrack()函数将创建一个绘图列表,每个绘图由指定的基因markerGenes。此函数将为groupBy参数中的每个组绘制一条轨迹。
markerGenes <- c(
"CD34", #Early Progenitor
"GATA1", #Erythroid
"PAX5", "MS4A1", #B-Cell Trajectory
"CD14", #Monocytes
"CD3D", "CD8A", "TBX21", "IL7R" #TCells
)
p <- plotBrowserTrack(
ArchRProj = projHeme2,
groupBy = "Clusters",
geneSymbol = markerGenes,
upstream = 50000,
downstream = 50000
)
要绘制特定基因的轨迹,我们可以从列表中选择一个。
grid::grid.newpage()
grid::grid.draw(p$CD14)
使用该plotPDF()功能,我们可以在图列表中为每个基因位点保存多页PDF,而只有一页。
plotPDF(plotList = p,
name = "Plot-Tracks-Marker-Genes.pdf",
ArchRProj = projHeme2,
addDOC = FALSE, width = 5, height = 5)
7.7启动ArchRBrowser
scATAC-seq数据分析固有的一项挑战是在各组中观察到的染色质可及性的基因组跟踪水平可视化。传统上,轨迹可视化需要对scATAC-seq片段进行分组,创建基因组覆盖率大佬,并对该轨迹进行标准化以进行定量可视化。通常,最终用户使用基因组浏览器(例如WashU表观基因组浏览器,UCSC基因组浏览器或IGV浏览器)来可视化这些测序轨迹。该过程涉及使用多个软件,对细胞群的任何更改或添加更多样本都需要重新生成bigwig文件等,这可能会很耗时。
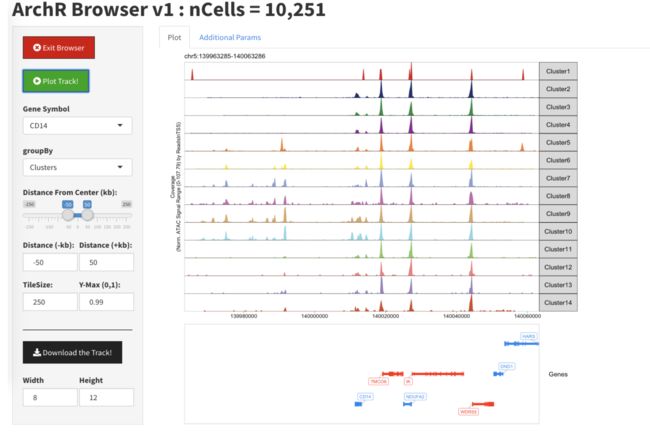
因此,ArchR具有基于Shiny的交互式基因组浏览器,该浏览器可以用一行代码启动ArchRBrowser(ArchRProj)。Arrow文件中实现的数据存储策略使该交互式浏览器可以动态更改单元分组,分辨率和规范化,从而实现实时轨道级别的可视化。ArchR基因组浏览器还创建PDF格式的高质量矢量化图像,以进行发布或分发。此外,浏览器GenomicRanges通过features参数接受用户提供的输入文件(例如用于显示特征的对象),或通过参数定义共同访问性,峰-基因链接或染色质构象数据循环的基因组交互文件loops。对于loops预期的格式是GRanges 对象,其起始位置代表一个环锚的中心位置,其终点位置代表另一环锚的中心位置。
要启动我们的本地交互式基因组浏览器,我们使用该ArchRBrowser()功能。
#ArchRBrowser(projHeme2)
开始时,我们将看到一个类似于以下的屏幕:

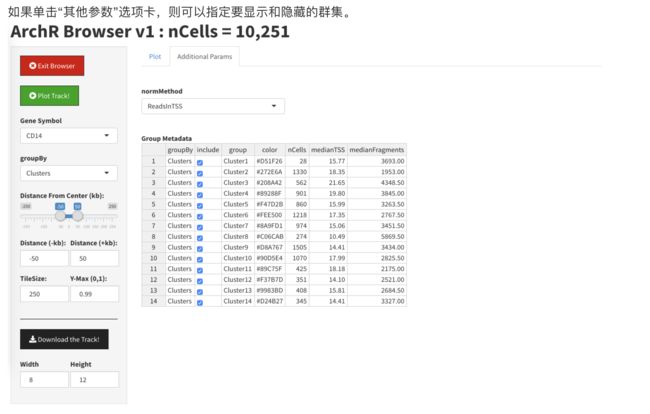
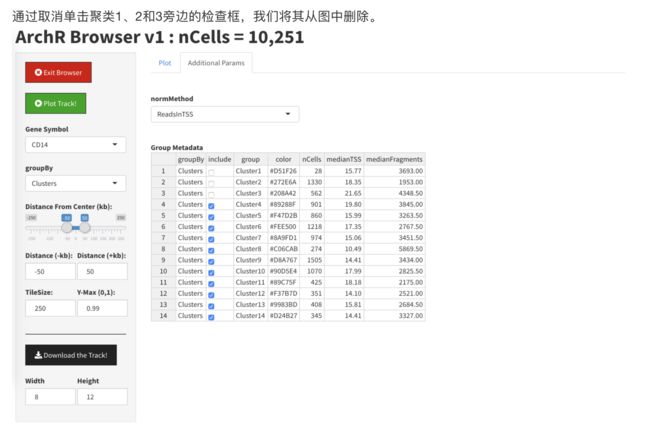
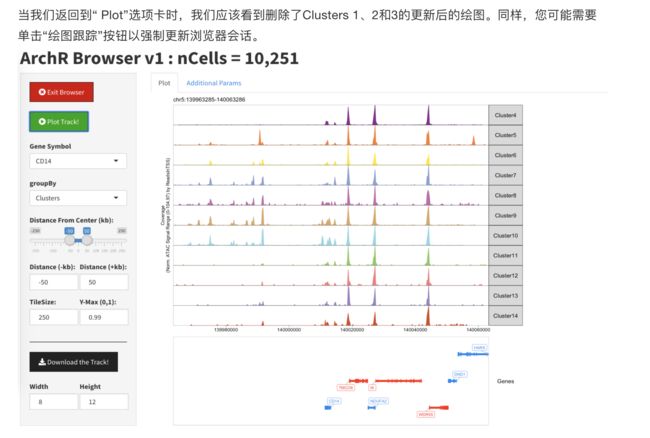
通过在“基因符号”框中选择一个基因,我们可以开始浏览。您可能需要单击“绘图跟踪”按钮以强制更新浏览器会话。

一旦我们绘制了基因座,我们就会看到一条轨迹代表数据中的每个簇。
参考材料:
https://www.archrproject.com/