刘小泽写于2020.7.21
为何取名叫“交响乐”?因为单细胞分析就像一个大乐团,需要各个流程的协同配合
单细胞交响乐1-常用的数据结构SingleCellExperiment
单细胞交响乐2-scRNAseq从实验到下游简介
单细胞交响乐3-细胞质控
单细胞交响乐4-归一化
单细胞交响乐5-挑选高变化基因
单细胞交响乐6-降维
单细胞交响乐7-聚类分群
单细胞交响乐8-marker基因检测
单细胞交响乐9-细胞类型注释
单细胞交响乐9-细胞类型注释
单细胞交响乐10-数据集整合后的批次矫正
单细胞交响乐11-多样本间差异分析
单细胞交响乐12-检测Doublet
单细胞交响乐13-细胞周期推断
单细胞交响乐14-细胞轨迹推断
单细胞交响乐15-scRNA与蛋白丰度信息结合
单细胞交响乐16-处理大型数据
单细胞交响乐17-不同单细胞R包的数据格式相互转换
单细胞交响乐18-实战一 Smart-seq2
单细胞交响乐19-实战二 STRT-Seq
单细胞交响乐20-实战三 10X 未过滤的PBMC数据
单细胞交响乐21-实战三 批量处理并整合多个10X PBMC数据
单细胞交响乐22-实战五 CEL-seq2
单细胞交响乐23-实战六 CEL-seq
单细胞交响乐24-实战七 SMARTer 胰腺细胞
单细胞交响乐25-实战八 Smart-seq2 胰腺细胞
单细胞交响乐26-实战九 胰腺细胞数据整合
单细胞交响乐27-实战十 CEL-seq-小鼠造血干细胞
单细胞交响乐28-实战十一 Smart-seq2-小鼠造血干细胞
单细胞交响乐29-实战十二 10X 小鼠嵌合体胚胎
1 前言
前面的种种都是作为知识储备,但是不实战还是记不住前面的知识
这是第十三个实战练习
数据来自Bach et al. (2017),使用的是10X的妊娠期小鼠乳腺上皮细胞
数据准备
library(scRNAseq)
sce.mam <- BachMammaryData(samples="G_1")
sce.mam
# class: SingleCellExperiment
# dim: 27998 2915
# metadata(0):
# assays(1): counts
# rownames: NULL
# rowData names(2): Ensembl Symbol
# colnames: NULL
# colData names(3): Barcode Sample Condition
# reducedDimNames(0):
# altExpNames(0):
数据初探
# 样本信息
sapply(names(colData(sce.mam)), function(x) head(colData(sce.mam)[,x]))
# Barcode Sample Condition
# [1,] "AAACCTGAGGATGCGT-1" "G_1" "Gestation"
# [2,] "AAACCTGGTAGTAGTA-1" "G_1" "Gestation"
# [3,] "AAACCTGTCAGCATGT-1" "G_1" "Gestation"
# [4,] "AAACCTGTCGTCCGTT-1" "G_1" "Gestation"
# [5,] "AAACGGGCACGAAATA-1" "G_1" "Gestation"
# [6,] "AAACGGGCAGACGCTC-1" "G_1" "Gestation"
ID转换
依然是整合行名 + 添加染色体信息
library(scater)
rownames(sce.mam) <- uniquifyFeatureNames(
rowData(sce.mam)$Ensembl, rowData(sce.mam)$Symbol)
library(AnnotationHub)
ens.mm.v97 <- AnnotationHub()[["AH73905"]]
rowData(sce.mam)$SEQNAME <- mapIds(ens.mm.v97, keys=rowData(sce.mam)$Ensembl,
keytype="GENEID", column="SEQNAME")
# 总共有13个线粒体基因
sum(grepl("MT",rowData(sce.mam)$SEQNAME))
# [1] 13
2 质控
依然是备份一下,把unfiltered数据主要用在质控的探索上
unfiltered <- sce.mam
使用线粒体信息进行过滤
is.mito <- rowData(sce.mam)$SEQNAME == "MT"
stats <- perCellQCMetrics(sce.mam, subsets=list(Mito=which(is.mito)))
qc <- quickPerCellQC(stats, percent_subsets="subsets_Mito_percent")
colSums(as.matrix(qc))
## low_lib_size low_n_features high_subsets_Mito_percent
## 0 0 143
## discard
## 143
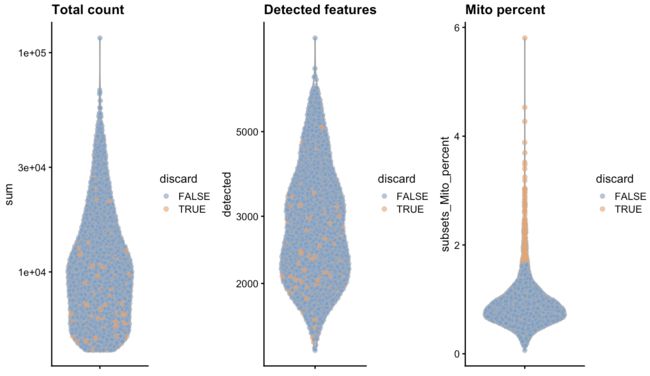
作图
colData(unfiltered) <- cbind(colData(unfiltered), stats)
unfiltered$discard <- qc$discard
gridExtra::grid.arrange(
plotColData(unfiltered, y="sum", colour_by="discard") +
scale_y_log10() + ggtitle("Total count"),
plotColData(unfiltered, y="detected", colour_by="discard") +
scale_y_log10() + ggtitle("Detected features"),
plotColData(unfiltered, y="subsets_Mito_percent",
colour_by="discard") + ggtitle("Mito percent"),
ncol=3
)
再看看线粒体含量与文库大小的关系
plotColData(unfiltered, x="sum", y="subsets_Mito_percent",
colour_by="discard") + scale_x_log10()

最后过滤
dim(unfiltered);dim(sce.mam)
# [1] 27998 2915
# [1] 27998 2772
3 归一化
使用去卷积的方法
library(scran)
set.seed(101000110)
clusters <- quickCluster(sce.mam)
sce.mam <- computeSumFactors(sce.mam, clusters=clusters)
sce.mam <- logNormCounts(sce.mam)
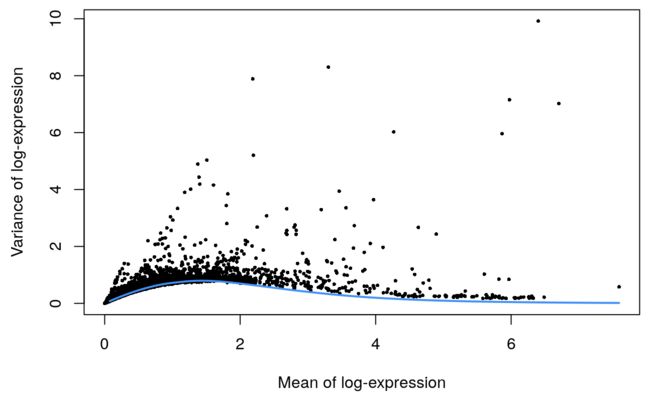
4 找高变异基因
这里由于是10X的数据,所以会有UMI信息,因此可以用基于泊松分布的模型构建方法
set.seed(00010101)
dec.mam <- modelGeneVarByPoisson(sce.mam)
top.mam <- getTopHVGs(dec.mam, prop=0.1)
最后做个图
plot(dec.mam$mean, dec.mam$total, pch=16, cex=0.5,
xlab="Mean of log-expression", ylab="Variance of log-expression")
curfit <- metadata(dec.mam)
curve(curfit$trend(x), col='dodgerblue', add=TRUE, lwd=2)
5 降维聚类
降维
library(BiocSingular)
set.seed(101010011)
sce.mam <- denoisePCA(sce.mam, technical=dec.mam, subset.row=top.mam)
sce.mam <- runTSNE(sce.mam, dimred="PCA")
# 检查PC的数量
ncol(reducedDim(sce.mam, "PCA"))
## [1] 15
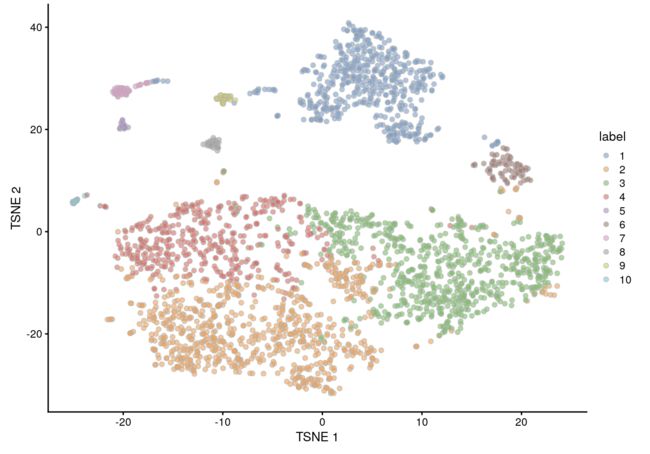
聚类
有一个很重要的参数是
k,含义是:the number of nearest neighbors used to construct the graph。如果k设置越大,得到的图之间联通程度越高,cluster也越大。因此这个参数也是可以不断尝试的
我们这里由于细胞数量比较多,所以设置的k就比较大,得到的cluster就少而大
snn.gr <- buildSNNGraph(sce.mam, use.dimred="PCA", k=25)
colLabels(sce.mam) <- factor(igraph::cluster_walktrap(snn.gr)$membership)
table(colLabels(sce.mam))
##
## 1 2 3 4 5 6 7 8 9 10
## 550 799 716 452 24 84 52 39 32 24
最后作图
plotTSNE(sce.mam, colour_by="label")
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
