首先要知道alevin属于salmon的一个工具,用于定量单细胞转录本,使用的比对方式是类似于kallisto的pseudo align,即不将reads比对到基因组上,该算法着重于确定一个 read 属于哪一个基因,而不关心这个 read 在基因上的位置。(Kallisto: 一个RNA-seq数据快速量化软件 http://blog.sciencenet.cn/blog-656335-984247.html;Kallisto ‘Pseudoalignment’ 原理:https://tinyheero.github.io/2015/09/02/pseudoalignments-kallisto.html)
因此最后没法生存单细胞比对的bam文件,这个bam文件还是有很多用处的,比如看一看单细胞中的突变位点情况。虽然alevin用下来确实很快,但是,不符合我的分析要求~
alevin的参考文档:
https://salmon.readthedocs.io/en/latest/alevin.html
使用Salmon对单细胞转录本进行定量
下载必要的文件
#### 仅包括蛋白编码的转录本
wget -nv ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_35/gencode.v35.pc_transcripts.fa.gz
#### 包括全部转录本
wget -nv ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_35/gencode.v35.transcripts.fa.gz
下载基因组注释gtf
wget -nv ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_35/gencode.v35.primary_assembly.annotation.gtf.gz
生成ENST到ENSG的匹配
bioawk -c gff '$feature=="transcript" {print $group}' <(gunzip -c gencode.v35.primary_assembly.annotation.gtf.gz) | awk -F ' ' '{print substr($4,2,length($4)-3) "\t" substr($2,2,length($2)-3)}' - > txp2gene.tsv
生成ENST到Gene Symbol的匹配
bioawk -c gff '$feature=="transcript" {print $group}' <(gunzip -c gencode.v35.primary_assembly.annotation.gtf.gz) | awk -F ' ' '{print substr($4,2,length($4)-3) "\t" substr($8,2,length($8)-3)}' > txp2gene_symbol.tsv
生成salmon的索引序列
./salmon-latest_linux_x86_64/bin/salmon index \
-i transcript_index \
-k 31 \
--gencode \
-p 4 \
-t gencode.v35.transcripts.fa.gz
运行单细胞转录本定量工具Alevin
./salmon-latest_linux_x86_64/bin/salmon alevin -l ISR \
-1 Sample_L001_R1_001.fastq.gz Sample_L002_R1_001.fastq.gz Sample_L003_R1_001.fastq.gz Sample_L004_R1_001.fastq.gz Sample_L005_R1_001.fastq.gz Sample_L006_R1_001.fastq.gz Sample_L007_R1_001.fastq.gz \
-2 Sample_L001_R2_001.fastq.gz Sample_L002_R2_001.fastq.gz Sample_L003_R2_001.fastq.gz Sample_L004_R2_001.fastq.gz Sample_L005_R2_001.fastq.gz Sample_L006_R2_001.fastq.gz Sample_L007_R2_001.fastq.gz \
--dropseq \
-i transcript_index \
-p 10 \
-o transcripts_output \
--tgMap txp2gene_symbol.tsv \
--expectCells 8000
速度确实很快,dropseq alignment protocol需要1天才能完成,alevin花了1个小时。
但是定量结果的差异还是很大:
dropseq alignment protocol最后比对出了8000个左右的细胞,
alevin比对出4300个细胞(其中还包括1000个 low confidence),这个原因下文会初步探索一下。
dropseq alignment protocol利用STAR 2-pass mode比对率在80%,
alevin比对率45%左右,这一点在Alevin github issue中有人提问过,开发者给出的回答是:STAR(或Hisat2)给出的比对率是全基因组的比对率,比对上的reads不一定是转录本,而alevin的比对率是reads比对至基因组转录本的比率。
差异可能来自于:
- 转录本的比对和技术策略:比对和“假比对”的技术差异
- alevin对barcode和umi有比较好的校正算法,drop-seq protocol的校正比较简单
- 选择的参考序列和注释文件的差异:drop-seq protocol使用全基因组序列建立索引,而alevin仅使用转录本序列
- 其他原因
后续的探索
比较一下alevin和drop-seq protocol的比对结果
alevin_result <- data.table::fread(input = "featureDump_alevin_result_20200929.txt")
# 读入drop-seq protocol计数的矩阵
p <- data.table::fread("./CH4-LN_hg38_gene_exon_tagged_CODING_UTR_INTRONIC.dge.txt.gz", data.table = F)
# alevin计数到的细胞中有98%是在drop-seq protocol中的
sum(alevin_result$CB %in% colnames(p))/nrow(alevin_result);rm(p)
# 取两次比对结果的交集
p <- FetchData(All, c("nCount_RNA","nFeature_RNA")) %>%
tibble::rownames_to_column("CB") %>%
inner_join(.,alevin_result, by = "CB")
library(ggplot2)
library(grid)
library(gridExtra)
library(ggpubr)
# 比较两个数据集reads数的比对结果
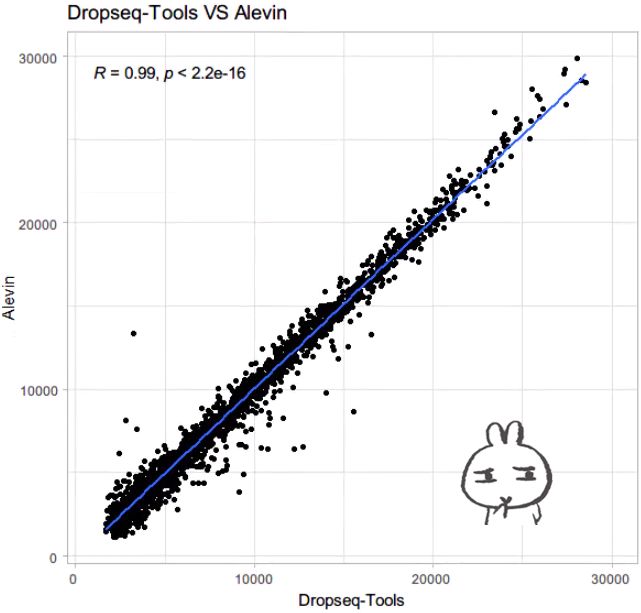
fig1 <- ggplot(p, aes(x=nCount_RNA, y=DeduplicatedReads)) +
geom_point() +
ggtitle("Dropseq-Tools VS Alevin") +
geom_smooth(method=lm, se=FALSE) +
scale_x_continuous(name = "Dropseq-Tools", limits = c(1E3,3E4)) + #, breaks = seq(5, 15, 2)
scale_y_continuous(name = "Alevin", limits = c(1E3,3E4)) + # , breaks = seq(5, 15, 2)
# annotation_custom(grob1) +
theme(plot.title = element_text(hjust = 0.5)) +
stat_cor(method="pearson") +
theme_light()
fig1
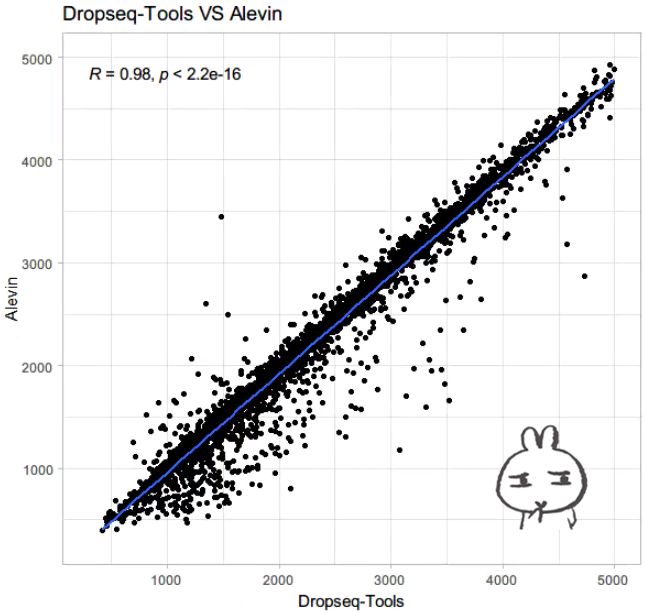
# 比较两个数据集基因数的比对结果
fig2 <- ggplot(p, aes(x=nFeature_RNA, y=NumGenesExpressed)) +
geom_point() +
ggtitle("Dropseq-Tools VS Alevin") +
geom_smooth(method=lm, se=FALSE) +
scale_x_continuous(name = "Dropseq-Tools", limits = c(300,5E3)) + #, breaks = seq(5, 15, 2)
scale_y_continuous(name = "Alevin", limits = c(300,5E3)) + # , breaks = seq(5, 15, 2)
# annotation_custom(grob1) +
theme(plot.title = element_text(hjust = 0.5)) +
stat_cor(method="pearson") +
theme_light()
fig2
# 看在Alevin中比对得到的细胞在Dropseq-Tools数据中的分布情况
All$inAlevin <- (Cells(All) %in% alevin_result$CB)
DimPlot(All, group.by = "inAlevin")
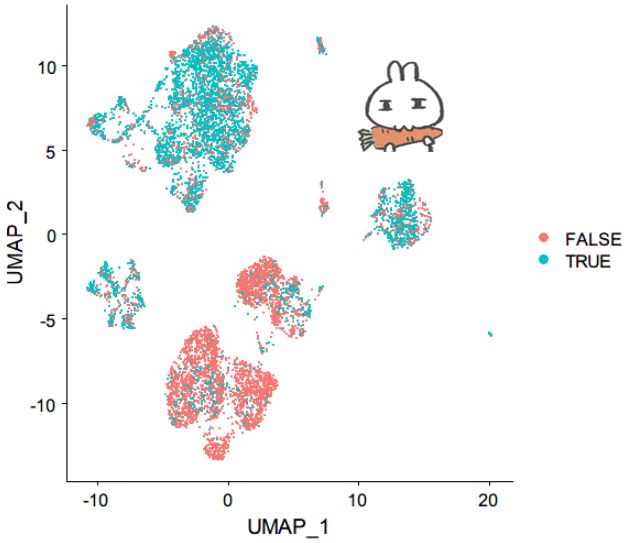
发现大部分的B细胞和T细胞没有被鉴定出,而大部分的肿瘤细胞,浆细胞,单核细胞被识别
这一点在alevin的issue (https://github.com/COMBINE-lab/salmon/issues/396) 中也提及:
"I think the CB frequency is most probably a bimodal distribution."
(可能是由于CB的双相分布导致alevin对细胞CB的错误估计)
确实B和T细胞的文库要偏小,或许和细胞大小相关?
结论:
- alevin非常快,非常非常快。虽然实际分析还是用传统比对流程好一些,但是快速的定量在多个数据集的探索,或者数据集质控方面还是会有一些优势。
- alevin在比对的结果方面和Dropseq-Tools一致。
- alevin可能会错误估计CB。
- 没有看到alevin对传统比对-定量流程的明显优势,本来希望alevin在barcode和umi的错误修复上会有优势,目前看来优势不明显。
- 目前还是倾向于选择传统的Dropseq-tools,生成的bam文件同其他pipeline整合(如mutation calling)也是很方便的。