scATAC分析神器ArchR初探-简介(1)
scATAC分析神器ArchR初探-ArchR进行doublet处理(2)
scATAC分析神器ArchR初探-创建ArchRProject(3)
scATAC分析神器ArchR初探-使用ArchR降维(4)
scATAC分析神器ArchR初探--使用ArchR进行聚类(5)
scATAC分析神器ArchR初探-单细胞嵌入(6)
scATAC分析神器ArchR初探-使用ArchR计算基因活性值和标记基因(7)
scATAC分析神器ArchR初探-scRNA-seq确定细胞类型(8)
scATAC分析神器ArchR初探-ArchR中的伪批次重复处理(9)
scATAC分析神器ArchR初探-使用ArchR-peak-calling(10)
scATAC分析神器ArchR初探-使用ArchR识别标记峰(11)
scATAC分析神器ArchR初探-使用ArchR进行主题和功能丰富(12)
scATAC分析神器ArchR初探-利用ArchR丰富ChromVAR偏差(13)
scATAC分析神器ArchR初探-使用ArchR进行足迹(14)
scATAC分析神器ArchR初探-使用ArchR进行整合分析(15)
scATAC分析神器ArchR初探-使用ArchR进行轨迹分析(16)
13-利用ArchR丰富ChromVAR偏差
如前几章所示,TF基序富集可以帮助我们预测哪些调控因子在我们感兴趣的细胞类型中最活跃。然而,这些富集不是基于每个细胞计算的,并且它们没有考虑到Tn5转座酶的插入序列偏差。chromVAR是Greenlead Lab打包的R,它是为解决这些问题而创建的。chromVAR用于根据稀疏染色质可及性数据预测每个细胞的TF活性富集。chromVAR的两个主要输出是:
- “偏差”-偏差是对给定特征(即基序)的每个单元可访问性与所有单元格或样本均值的预期可访问性相差多少的偏差校正度量。
- “ z分数”-z分数(也称为“偏差分数”)是所有像元上每个偏差校正偏差的z分数。偏差评分的绝对值与每个单元的读取深度相关。这是因为,随着阅读次数的增加,给定特征(即基序)在每个单元可访问性方面与预期的差异要比偶然发生的差异更有把握。
chromVAR的主要局限性之一是它是在scATAC-seq数据生成的早期阶段设计的,当时实验由数百个细胞组成。在这种实验规模下,chromVAR可以轻松地将整个逐峰矩阵读取到内存中,以快速计算TF偏差。但是,当前的实验方法使用了成千上万个单元,生成了逐个单元的矩阵,这些矩阵很难读入内存。即使对于大小适中的50,000个数据集,这也会导致运行时间和内存使用量急剧增加。
为了避免这些限制,ArchR通过独立分析样本子矩阵来实施相同的chromVAR分析工作流程。
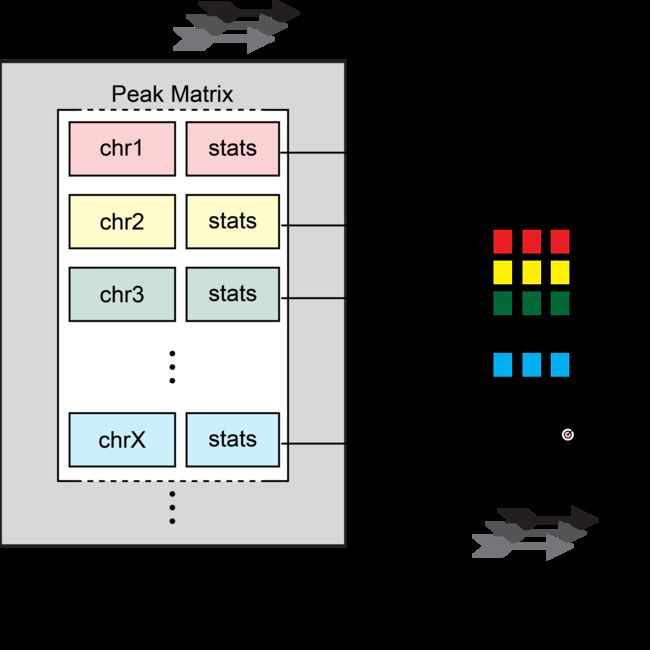
首先,ArchR读取每个子样本中所有单元格上每个峰的全局可访问性。其次,对于每个峰,ArchR都会识别一组与GC含量和可及性相匹配的背景峰。第三,ArchR使用峰的背景设置和全局可访问性来独立计算每个样本的带有chromVAR的偏差校正偏差。此实现要求在任何给定时间仅将5,000-10,000个单元中的数据加载到内存中,从而最大程度地减少内存需求,使用chromVAR进行可扩展的分析并提高运行时性能。
13.1主题偏差
首先,请确保已在中添加了主题注释ArchRProject。
if("Motif" %ni% names(projHeme5@peakAnnotation)){
projHeme5 <- addMotifAnnotations(ArchRProj = projHeme5, motifSet = "cisbp", name = "Motif")
}
我们还需要添加一组背景峰,用于计算偏差。使用该chromVAR::getBackgroundPeaks()函数选择背景峰,该函数根据GC含量的相似性和所有样品之间的碎片数(使用马氏距离)对峰进行采样。
projHeme5 <- addBgdPeaks(projHeme5)
现在,我们准备使用该addDeviationsMatrix()函数计算所有图案注释中每个单元的偏差。该函数有一个可选参数,名为matrixName,它使我们能够定义将存储在Arrow文件中的偏差矩阵的名称。如果我们没有为该参数提供值,如下面的示例所示,此函数通过在的名称上添加单词“ Matrix”来创建矩阵名称peakAnnotation。下面的示例在我们每个名为“ MotifMatrix”的Arrow文件中创建一个偏差矩阵。
projHeme5 <- addDeviationsMatrix(
ArchRProj = projHeme5,
peakAnnotation = "Motif",
force = TRUE
)
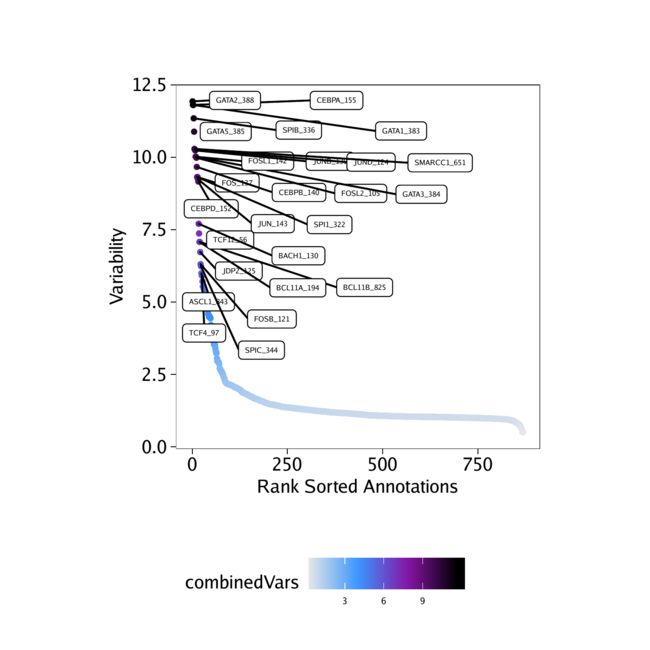
要访问这些偏差,我们使用getVarDeviations()函数。如果我们希望该函数返回一个ggplot对象,则进行设置,plot = TRUE否则,该函数将返回该DataFrame对象。该head说的DataFrame对象是默认运行该功能时显示。
plotVarDev <- getVarDeviations(projHeme5, name = "MotifMatrix", plot = TRUE)
从的上述快照中DataFrame,您可以看到seqnames的MotifMatrix不是染色体。通常情况下,像矩阵TileMatrix,PeakMatrix和GeneScoreMatrix我们保存在染色体上的信息seqnames。在MotifMatrix不具有任何相应的位置上的信息,但,相反,同时存储从chromVAR了“devations”和“z分数”到使用两个不同seqnames相同的基质- deviations和z。稍后,如果您尝试在这类函数中使用MotifMatrix(属于类Sparse.Assays.Matrix),这一点就变得很重要getMarkerFeatures()。在这些类型的操作中,ArchR会希望您将其子集化为MotifMatrix两者之一seqnames(即,选择z得分或偏差以执行计算)。
然后,我们可以绘制这些变量偏差。
plotVarDev
要保存此图的可编辑矢量化版本,我们使用plotPDF()函数。
plotPDF(plotVarDev, name = "Variable-Motif-Deviation-Scores", width = 5, height = 5, ArchRProj = projHeme5, addDOC = FALSE)
如果我们想提取一个基序子集进行下游分析该怎么办?我们可以使用getFeatures()函数来做到这一点。paste(motifs, collapse="|")下面的语句创建了一个连接or语句,该语句可以选择所有主题。
motifs <- c("GATA1", "CEBPA", "EBF1", "IRF4", "TBX21", "PAX5")
markerMotifs <- getFeatures(projHeme5, select = paste(motifs, collapse="|"), useMatrix = "MotifMatrix")
markerMotifs
如上所述,MotifMatrix包含seqnamesz得分和偏差,如上用“ z:”和“偏差:”所示。要仅获取与z得分相对应的特征,我们可以使用grep。不幸的是,在上面显示的示例主题中,您可以看到,除了“ EBF1”之外,我们还选择了“ SREBF1”,我们不想对其进行分析。因此,我们在下面使用%ni%表达式(它是ArchR辅助函数)将其删除,该函数提供与%in%R 的反义词。
markerMotifs <- grep("z:", markerMotifs, value = TRUE)
markerMotifs <- markerMotifs[markerMotifs %ni% "z:SREBF1_22"]
markerMotifs
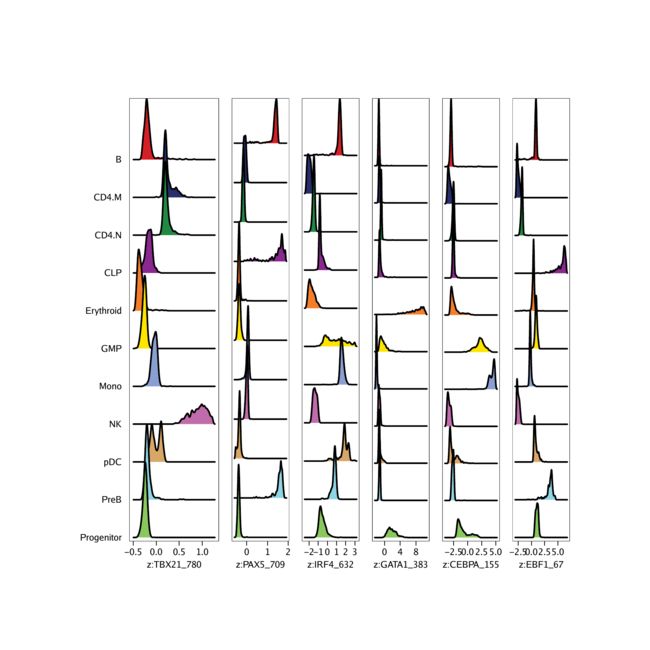
现在我们有了我们感兴趣的功能的名称,我们可以绘制每个群集的chromVAR偏差评分的分布。请注意,我们提供了我们在基因评分分析期间先前计算的估算权重。提醒一下,这些估算权重使我们能够使附近单元格上的信号平滑,这对于稀疏的scATAC-seq数据很有帮助。
p <- plotGroups(ArchRProj = projHeme5,
groupBy = "Clusters2",
colorBy = "MotifMatrix",
name = markerMotifs,
imputeWeights = getImputeWeights(projHeme5)
)
我们可以cowplot在一个图中绘制所有这些图案的分布。
p2 <- lapply(seq_along(p), function(x){
if(x != 1){
p[[x]] + guides(color = FALSE, fill = FALSE) +
theme_ArchR(baseSize = 6) +
theme(plot.margin = unit(c(0.1, 0.1, 0.1, 0.1), "cm")) +
theme(
axis.text.y=element_blank(),
axis.ticks.y=element_blank(),
axis.title.y=element_blank()
) + ylab("")
}else{
p[[x]] + guides(color = FALSE, fill = FALSE) +
theme_ArchR(baseSize = 6) +
theme(plot.margin = unit(c(0.1, 0.1, 0.1, 0.1), "cm")) +
theme(
axis.ticks.y=element_blank(),
axis.title.y=element_blank()
) + ylab("")
}
})
do.call(cowplot::plot_grid, c(list(nrow = 1, rel_widths = c(2, rep(1, length(p2) - 1))),p2))
要保存此图的可编辑矢量化版本,我们使用plotPDF()函数。
plotPDF(p, name = "Plot-Groups-Deviations-w-Imputation", width = 5, height = 5, ArchRProj = projHeme5, addDOC = FALSE)
无需查看这些z分数的分布,而是可以像我们之前对基因分数所做的那样,将z分数覆盖在我们的UMAP嵌入上。
p <- plotEmbedding(
ArchRProj = projHeme5,
colorBy = "MotifMatrix",
name = sort(markerMotifs),
embedding = "UMAP",
imputeWeights = getImputeWeights(projHeme5)
)
我们可以使用绘制所有这些图案UMAP cowplot。
p2 <- lapply(p, function(x){
x + guides(color = FALSE, fill = FALSE) +
theme_ArchR(baseSize = 6.5) +
theme(plot.margin = unit(c(0, 0, 0, 0), "cm")) +
theme(
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)
})
do.call(cowplot::plot_grid, c(list(ncol = 3),p2))
要查看这些TF偏差z分数与通过相应TF基因的基因得分推断的基因表达相比如何,我们可以在UMAP嵌入中叠加每个TF的基因得分。
markerRNA <- getFeatures(projHeme5, select = paste(motifs, collapse="|"), useMatrix = "GeneScoreMatrix")
markerRNA <- markerRNA[markerRNA %ni% c("SREBF1","CEBPA-DT")]
markerRNA
p <- plotEmbedding(
ArchRProj = projHeme5,
colorBy = "GeneScoreMatrix",
name = sort(markerRNA),
embedding = "UMAP",
imputeWeights = getImputeWeights(projHeme5)
)
p2 <- lapply(p, function(x){
x + guides(color = FALSE, fill = FALSE) +
theme_ArchR(baseSize = 6.5) +
theme(plot.margin = unit(c(0, 0, 0, 0), "cm")) +
theme(
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)
})
do.call(cowplot::plot_grid, c(list(ncol = 3),p2))
同样,由于我们之前已将scATAC-seq数据与相应的scRNA-seq数据链接在一起,因此我们可以在UMAP嵌入中绘制每个TF的链接基因表达。
markerRNA <- getFeatures(projHeme5, select = paste(motifs, collapse="|"), useMatrix = "GeneIntegrationMatrix")
markerRNA <- markerRNA[markerRNA %ni% c("SREBF1","CEBPA-DT")]
markerRNA
p <- plotEmbedding(
ArchRProj = projHeme5,
colorBy = "GeneIntegrationMatrix",
name = sort(markerRNA),
embedding = "UMAP",
continuousSet = "blueYellow",
imputeWeights = getImputeWeights(projHeme5)
)
p2 <- lapply(p, function(x){
x + guides(color = FALSE, fill = FALSE) +
theme_ArchR(baseSize = 6.5) +
theme(plot.margin = unit(c(0, 0, 0, 0), "cm")) +
theme(
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)
})
do.call(cowplot::plot_grid, c(list(ncol = 3),p2))
13.2 ArchR和自定义偏差
在“峰注释富集”一章中,我们介绍了如何为任何一组基因组区域创建峰注释。这包括(i)ArchR支持的区域集,例如来自ENCODE的精选TF结合位点和来自批量ATAC-seq的峰集,以及(ii)用户提供的自定义区域集。如果您尚未阅读本节,建议您这样做以更好地了解峰注释的工作方式。
这些峰注释可以与图案相同的方式用于偏差计算中。在这里,我们提供了有关如何进行这些分析的示例,但请注意,下游分析与上一节中有关主题的显示相同,因此,我们没有在代码的每个步骤中提供详尽的细节。在Arrow文件中创建偏差矩阵后,其余部分相同。
13.2.1编码TFBS
如果您尚未为“ EncodeTFBS”区域集添加注释矩阵,请立即执行。
if("EncodeTFBS" %ni% names(projHeme5@peakAnnotation)){
projHeme5 <- addArchRAnnotations(ArchRProj = projHeme5, collection = "EncodeTFBS")
}
然后,我们创建一个偏差矩阵,将此峰注释提供给peakAnnotation参数。
projHeme5 <- addDeviationsMatrix(
ArchRProj = projHeme5,
peakAnnotation = "EncodeTFBS",
force = TRUE
)
我们可以创建排名偏差的点图。
plotVarDev <- getVarDeviations(projHeme5, plot = TRUE, name = "EncodeTFBSMatrix")
plotVarDev
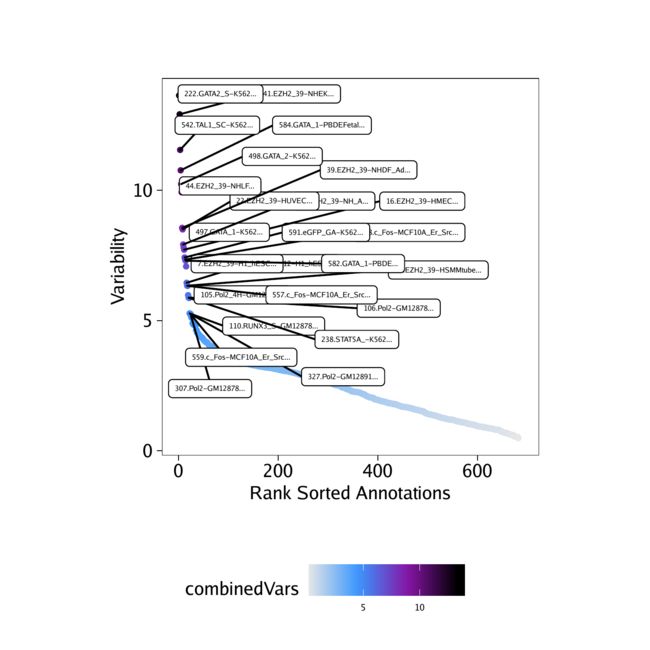
plotPDF(plotVarDev, name = "Variable-EncodeTFBS-Deviation-Scores", width = 5, height = 5, ArchRProj = projHeme5, addDOC = FALSE)
或者,我们可以将这些TF结合位点子集化为我们感兴趣的特定基序,然后在UMAP嵌入中绘制每个单元的偏差z分数。
tfs <- c("GATA_1", "CEBPB", "EBF1", "IRF4", "TBX21", "PAX5")
getFeatures(projHeme5, select = paste(tfs, collapse="|"), useMatrix = "EncodeTFBSMatrix")
markerTFs <- getFeatures(projHeme5, select = paste(tfs, collapse="|"), useMatrix = "EncodeTFBSMatrix")
markerTFs <- sort(grep("z:", markerTFs, value = TRUE))
TFnames <- stringr::str_split(stringr::str_split(markerTFs, pattern = "\\.", simplify=TRUE)[,2], pattern = "-", simplify = TRUE)[,1]
markerTFs <- markerTFs[!duplicated(TFnames)]
markerTFs
p <- plotEmbedding(
ArchRProj = projHeme5,
colorBy = "EncodeTFBSMatrix",
name = markerTFs,
embedding = "UMAP",
imputeWeights = getImputeWeights(projHeme5)
)
p2 <- lapply(p, function(x){
x + guides(color = FALSE, fill = FALSE) +
theme_ArchR(baseSize = 6.5) +
theme(plot.margin = unit(c(0, 0, 0, 0), "cm")) +
theme(
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)
})
do.call(cowplot::plot_grid, c(list(ncol = 3),p2))
13.2.2批量ATAC序列
同样,我们可以使用ArchR固化的批量ATAC-seq峰集进行图案偏差计算。
如果您尚未为“ EncodeTFBS”区域集添加注释矩阵,请立即执行。
if("ATAC" %ni% names(projHeme5@peakAnnotation)){
projHeme5 <- addArchRAnnotations(ArchRProj = projHeme5, collection = "ATAC")
}
然后,我们创建一个偏差矩阵,将此峰注释提供给peakAnnotation参数。
projHeme5 <- addDeviationsMatrix(
ArchRProj = projHeme5,
peakAnnotation = "ATAC",
force = TRUE
)
我们可以创建排名偏差的点图。
plotVarDev <- getVarDeviations(projHeme5, plot = TRUE, name = "ATACMatrix")
plotVarDev
plotPDF(plotVarDev, name = "Variable-ATAC-Deviation-Scores", width = 5, height = 5, ArchRProj = projHeme5, addDOC = FALSE)
或者,我们可以在UMAP嵌入中为每个像元绘制每个单元格的偏差z分数。
ATACPeaks <- c("Heme_HSC", "Heme_LMPP", "Heme_Ery", "Heme_Mono", "Heme_CD4", "Heme_CD8", "Heme_B", "Heme_NK", "IAtlas_DC_Plasmacytoid")
getFeatures(projHeme5, select = paste(ATACPeaks, collapse="|"), useMatrix = "ATACMatrix")
markerATAC <- getFeatures(projHeme5, select = paste(ATACPeaks, collapse="|"), useMatrix = "ATACMatrix")
markerATAC <- sort(grep("z:", markerATAC, value = TRUE))
markerATAC
p <- plotEmbedding(
ArchRProj = projHeme5,
colorBy = "ATACMatrix",
name = markerATAC,
embedding = "UMAP",
imputeWeights = getImputeWeights(projHeme5)
)
p2 <- lapply(p, function(x){
x + guides(color = FALSE, fill = FALSE) +
theme_ArchR(baseSize = 6.5) +
theme(plot.margin = unit(c(0, 0, 0, 0), "cm")) +
theme(
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)
})
do.call(cow
13.2.3自定义偏差
代替使用上述的ArchR固化区域集,我们可以提供自己的自定义区域集作为峰注解。这些自定义注释的使用方式与ArchR-curated注释的使用方式完全相同。
首先,如果您尚未在上一章中创建此“ EncodePeaks”注释,请通过下载一些ENCODE峰集并调用来创建它addPeakAnnotations()。
EncodePeaks <- c(
Encode_K562_GATA1 = "https://www.encodeproject.org/files/ENCFF632NQI/@@download/ENCFF632NQI.bed.gz",
Encode_GM12878_CEBPB = "https://www.encodeproject.org/files/ENCFF761MGJ/@@download/ENCFF761MGJ.bed.gz",
Encode_K562_Ebf1 = "https://www.encodeproject.org/files/ENCFF868VSY/@@download/ENCFF868VSY.bed.gz",
Encode_K562_Pax5 = "https://www.encodeproject.org/files/ENCFF339KUO/@@download/ENCFF339KUO.bed.gz"
)
if("ChIP" %ni% names(projHeme5@peakAnnotation)){
projHeme5 <- addPeakAnnotations(ArchRProj = projHeme5, regions = EncodePeaks, name = "ChIP")
}
然后,我们从该峰注释中制作一个偏差矩阵。
projHeme5 <- addDeviationsMatrix(
ArchRProj = projHeme5,
peakAnnotation = "ChIP",
force = TRUE
)
分析工作流程的其余部分与上面多次介绍的内容相同。
我们可以绘制排名偏差。
plotVarDev <- getVarDeviations(projHeme5, plot = TRUE, name = "ChIPMatrix")
plotVarDev
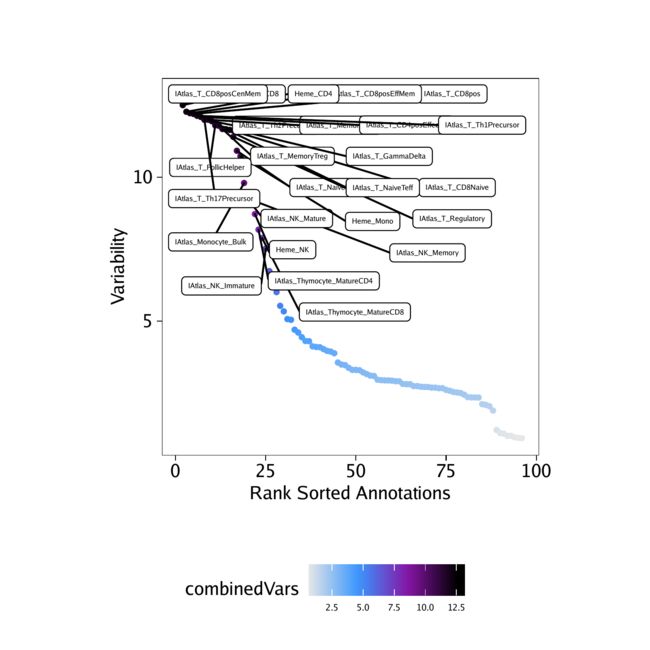
plotPDF(plotVarDev, name = "Variable-ChIP-Deviation-Scores", width = 5, height = 5, ArchRProj = projHeme5, addDOC = FALSE)
我们可以绘制覆盖在我们的UMAP嵌入中的偏差z得分。
markerChIP <- getFeatures(projHeme5, useMatrix = "ChIPMatrix")
markerChIP <- sort(grep("z:", markerChIP, value = TRUE))
markerChIP
p <- plotEmbedding(
ArchRProj = projHeme5,
colorBy = "ChIPMatrix",
name = markerChIP,
embedding = "UMAP",
imputeWeights = getImputeWeights(projHeme5)
)
p2 <- lapply(p, function(x){
x + guides(color = FALSE, fill = FALSE) +
theme_ArchR(baseSize = 6.5) +
theme(plot.margin = unit(c(0, 0, 0, 0), "cm")) +
theme(
axis.text.x=element_blank(),
axis.ticks.x=element_blank(),
axis.text.y=element_blank(),
axis.ticks.y=element_blank()
)
})
do.call(cowplot::plot_grid, c(list(ncol = 2),p2))
参考材料:
https://www.archrproject.com/