Seurat Weekly NO.0 || 开刊词
在过去的一周里Seurat社区在github总提问数由上周的3066上升到3090,当然有同一问题反复讨论的情况,也有之前的问题还有人再问的情况,总的来说上周其在github中的issues往来邮件一共182封.本次主要和大家分享一下几个,本期的封面故事是:
Resolution parameter in Seurat's FindClusters function for larger cell numbers
即,在做细胞分群的时候如何确定分群数据量?在这之前我们先来看几个有趣的问答吧。
Error using WhichCells/subset in a for loop
https://github.com/satijalab/seurat/issues/1053
https://github.com/satijalab/seurat/issues/1839
当我们需要循环地对某个基因列表用WhichCells/subset做条件筛选的时候:
genes <- c("KIF22", "RPGRIP1L")
v <- c(1:length(genes))
for (i in v){
cells <- WhichCells(seurat_object, expression = genes[i] > 0)
print(length(cells))
}
我们发现:
Error in FetchData(object = object, vars = expr.char[vars.use], cells = cells, : None of the requested variables were found:
而单独使用基因名是可以的:
WhichCells(seurat_object, expression = KIF22 > 0)
为什么呢?为什么基因名作为循环变量就不行了呢?我们发现循环的时候基因名是一个字符串,而单独写的时候并不是字符串(没加引号),GitHub的回答是:
Much like R's
subsetfunction,subset.Seuratis designed for interactive use only. While we currently don't offer a programmatic way to subset Seurat objects based on feature expression, this can be accomplished relatively easily using which andFetchData:
var1 <- "GeneName"
expr <- FetchData(object = object, vars = var1)
object[, which(x = expr > low & expr < high)]
s所以在我们设计循环体的时候,不能直接用WhichCells/subset,可以用上面的方式:
library(Seurat)
gene_cells = list()
items <-c( 'LGALS3', 'S100A11')
for (gene1 in items)
{
if (gene1 %in% rownames(pbmc_small))
{
expr <- FetchData(object = pbmc_small, vars = gene1)
gene_cells[[gene1]] <- pbmc_small[, which(x = expr >= 0.5)]
}
}
gene_cells
$LGALS3
An object of class Seurat
230 features across 24 samples within 1 assay
Active assay: RNA (230 features)
2 dimensional reductions calculated: pca, tsne
$S100A11
An object of class Seurat
230 features across 45 samples within 1 assay
Active assay: RNA (230 features)
2 dimensional reductions calculated: pca, tsne
关于Seurat的subset还有一个诡异的地方:
pbmc_small[VariableFeatures(object = pbmc_small), ]
pbmc_small[, 1:10]
subset(x = pbmc_small, subset = MS4A1 > 4)
subset(x = pbmc_small, subset = `DLGAP1-AS1` > 2)
subset(x = pbmc_small, idents = '0', invert = TRUE)
subset(x = pbmc_small, subset = MS4A1 > 3, slot = 'counts')
subset(x = pbmc_small, features = VariableFeatures(object = pbmc_small))
当基因名中有-的时候,需要用反单引号引起来才能行。
Fixing batch effect with seurat integrate
biostars : https://www.biostars.org/p/390189/
上一期我们讨论了如何在Seurat分析多个样本。今天选择这个问题的原因是它让我们明白在使用工具的时候,对工具原理的了解是多么重要。在运用概念时,对概念之前的关系理解是多么重要。
提问者拿出nFeatures_RNA 的箱型图,问这是不是批次效应造成的,这能不能说明样本间有批次效应?

这是一个经典的问题,类似地我们可以拿出两个样本间任何有差异的东西来做出这样的提问: 这是不是批次效应造成的,这能不能说明样本间有批次影响?那么,批次效应真的是万恶之源了。其实,在观察到差异的时候,我们首先要问的是:为什么。然后观察造成这种差异的原因,也许批次效应是最后应该考虑的。就拿这个问题来说吧,箱型图说明不同处理间细胞捕获的基因数量不同,那么是哪些基因不同呢可不可以做一个overlap,看看共有的基因是哪些,哪些又是缺失的?只有阐明了差异的来源才能想办法消除。
Resolution parameter in Seurat's FindClusters function for larger cell numbers
bioinformatics : https://bioinformatics.stackexchange.com/questions/4297/resolution-parameter-in-seurats-findclusters-function-for-larger-cell-numbers
github : https://github.com/satijalab/seurat/issues/1565
我的细胞到底分多少个群是合适的?这是一个广泛而经典问题。就单细胞技术而言,我们常说每个细胞都是不同的,也就是说你总可以分到最细以单细胞为单位,但是这样就失去高通量的意义了。在低通量下,我们可以着眼于单个细胞,现在成千上万的细胞,一个一个看是不切实际的。那么,我的细胞到底分多少个群是合适的?
这个问题表现在Seurat中就是:Finding optimal cluster resolution in Seurat 3? 我们知道,不同的resolution参数会带来不同的分群结果。先看一下github上面的回答:
While Seurat doesn't have tools for comparing cluster resolutions, there is a tool called clustree designed for this task and works on
Seuratv3 objects natively. It's available on CRAN and can be installed with a simpleinstall.packages('clustree')
clustree我们之前讲过,可以全局地查看不同分群结果:
#先执行不同resolution 下的分群
library(Seurat)
pbmc_small <- FindClusters(
object = pbmc_small,
resolution = c(seq(.4,1.6,.2))
)
clustree([email protected], prefix = "RNA_snn_res.")
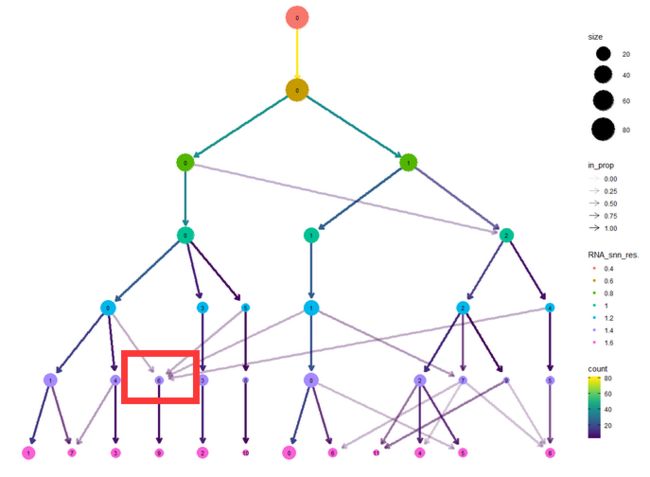
在clustree的图中我们看到不同resolution的取值情况下分群的关系。既然我们最终是以群为单位来分析的,我们肯定是希望每个群是比较纯的。如图可以看到在倒数第二层级有个亚群来自不同的分群,这有可能是:
- 分群过度,把原来分群的中应有的异质性也提炼出来单独作为一群了
- 上一层级分群不足,还包含了不该有的异质性。
这里就带来灵魂拷问了,就拿B细胞来说吧,它本身也是有异质性的啊,那么他的异质性是如何的呢?我们知道,某一类细胞内的异质性一般是要小于细胞群之间的异质性的。所以,拿到这个图我们就可以根据自己带着生物学意义的期望来做一个判断了。
其实,我们也知道分群终究是非监督的,只是数据驱动的,并不掺杂着数据(表达谱)以外的生物学意义。如果抛开这些生物学意义,其实是有一些办法来评价分群结果的:

这些方法也是在做群内和群之间的比较,得出类似群纯度的度量单位来评价分群结果。在不久前张泽民老师团队的一篇文章中提到过一种方法:ROGUE: an entropy-based universal metric for assessing the purity of single cell population。
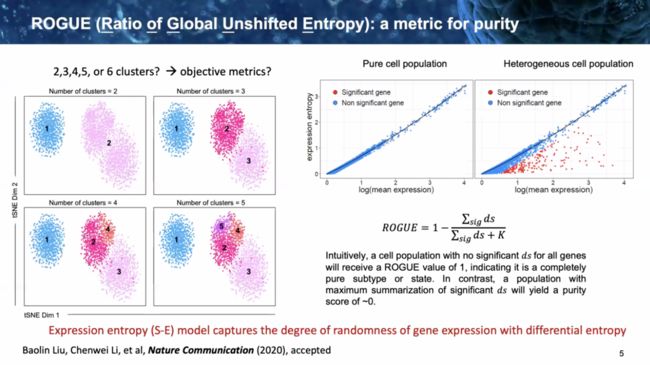
该方法已被封装为一个R包: https://github.com/PaulingLiu/ROGUE
我们看到已经有不少的方法来做分群的评估了,还有:IKAP—Identifying K mAjor cell Population groups in single-cell RNA-sequencing analysis :
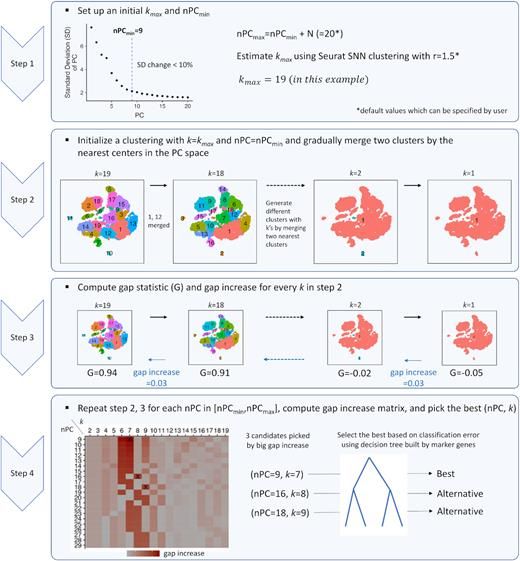
以上这些方法大同小异,核心的问题是,或者研究者真正关心的是:
哪种分群结果的生物解释性高?
正所谓:分析总会有结果,看你敢用不敢用。