基因组重测序数据目的:需要检测基因组中的变异,找到并定位这些突变位点
条件:参考基因组、重测序数据、
分析流程:

Step 1 下载sra文件
使用软件:Aspera
Aspera用法:ascp [参数] 目标文件 保存路径
步骤:
1、建立Seq文件夹(在home目录下)
mkdir ~/Seq
2、下载
ascp -T -i /home/raewyn/.aspera/connect/etc/asperaweb_id_dsa.openssh -k 1 -l 200m [email protected]:/sra/sra-instant/reads/ByRun/sra/SRR/SRR194/SRR1947852/SRR1947852.sra ~/Seq/
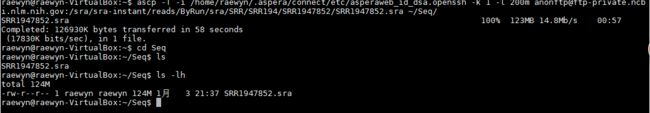
参数详解:
-T 不进行加密。若不添加此参数,可能会下载不了。
-i string 输入私钥,安装 aspera 后有在目录 ~/.aspera/connect/etc/ 下有几个私钥,
使用 linux 服务器的时候一般使用 asperaweb_id_dsa.openssh 文件作为私钥。
-k 1 支持断点续传
-l string 设置最大传输速度,比如设置为 200M 则表示最大传输速度为 200m/s。
若不设置该参数,则一般可达到10m/s的速度,而设置了,传输速度可以更高。
FTP下载地址:ftp://ftp.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR194/SRR1947852/SRR1947852.sra
改为[email protected]:/sra/sra-instant/reads/ByRun/sra/SRR/SRR194/SRR1947852/SRR1947852.sra
Step 2 解压SRA文件为fastq
语法:fastq-dump --split-files 文件名或 fastq-dump --gzip --split-files 文件名(解压为gz文件,节省空间)
即:fastq-dump --gzip --split-files SRR1947852.sra
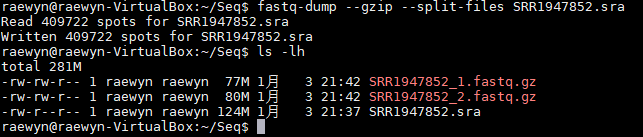
双端测序一般有两个文件(也可通过某种规则把两个文件合并成一个)。第一个文件与第二个文件的行数完全一样,且测序序列的排列顺序完全一致。
在第一个文件中,描述信息的结尾是“/1”,表示是双端测序的一端;第二个文件中同样位置/行数的相对应的测序序列的描述信息则以“/2”结尾,表示是双端测序的另一端 。
Step 3 去接头,低质量碱基
使用软件:fastqc
格式:fastqc 【处理的文件文件名】
步骤:
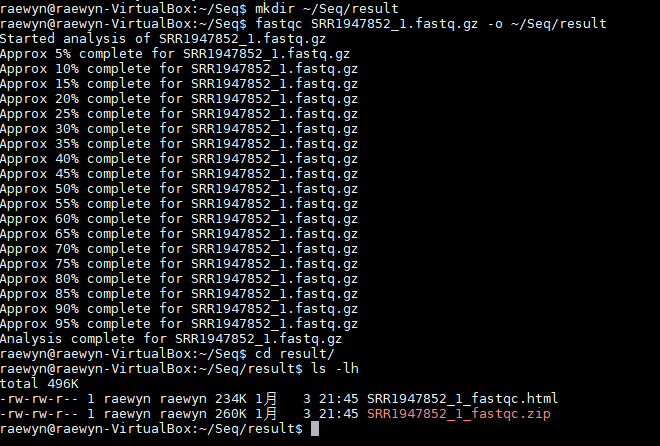
1、建立一个放置结果的文件夹(在Seq目录下)
mkdir ~/Seq/result
2、用fastqc去接头及低质量碱基
fastqc SRR1947852_1.fastq.gz -o ~/Seq/result
-o 指定生成到某个文件夹
3、生成了一个fastqc.html和一个fastqc.zip文件
Step 4 比对
Reads比对到参考基因组
使用软件:bwa
语法:
bwa index 参考基因组(建立索引) ,
bwa 参考基因组 处理文件 > 比对后的文件名( bwa的使用需要两种输入文件:①索引的Reference genome data(fasta格式 .fa, .fasta, .fna)②Short reads data (fastaq格式 .fastaq, .fq))
步骤:
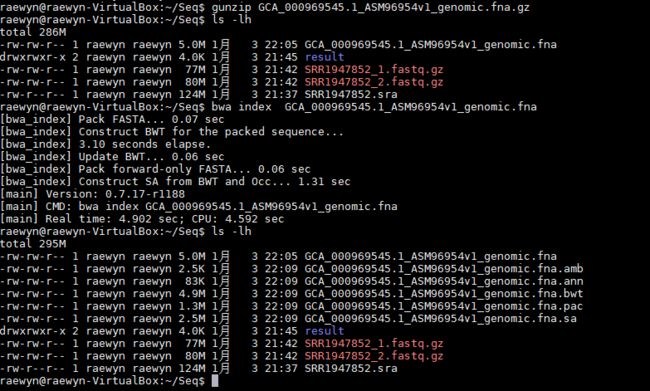
1、解开压缩包
gunzip GCA_000969545.1_ASM96954v1_genomic.fna.gz
2、bwa index建立索引(在bwa_test目录下)
bwa index GCA_000969545.1_ASM96954v1_genomic.fna
3、reads比对到参考序列(比对后的文件为 )
bwa mem GCA_000969545.1_ASM96954v1_genomic.fna SRR1947852_1.fastq.gz SRR1947852_2.fastq.gz > seq_sam_1947852.sam
即可得到一个sam文件
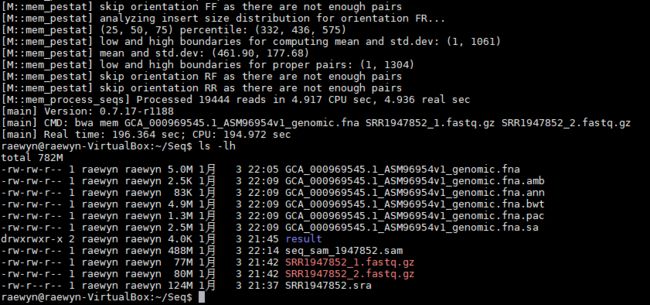
Step 5 排序索引后的比对结果 (将sam转为bam格式)
使用软件:samtools
语法:samtools view [options]| [region1 [...]]
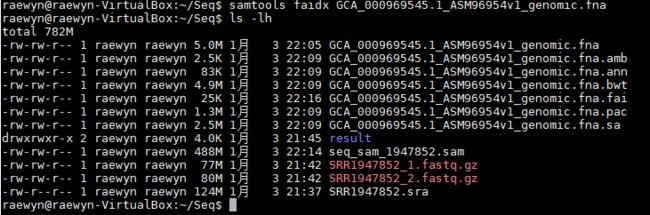
1、为参考基因组建立索引,生成了prefix.fai文件
samtools faidx GCA_000969545.1_ASM96954v1_genomic.fna
语法详解:
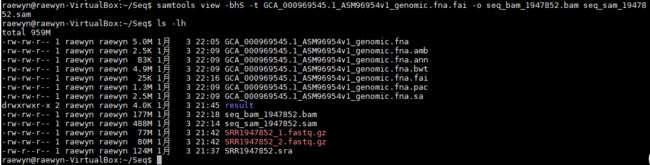
2、sam文件转为bam文件
samtools view -bhS -t GCA_000969545.1_ASM96954v1_genomic.fna.fai -o seq_bam_1947852.bam seq_sam_1947852.sam
语法详解:
-b output BAM 默认下输出是 SAM 格式文件,该参数设置输出 BAM 格式
-h print header for the SAM output 默认下输出的 sam 格式文件不带 header,该参数设定输出
sam文件时带 header 信息
-S input is SAM 默认下输入是 BAM 文件,若是输入是 SAM 文件,则最好加该参数,否则有
时候会报错
-T FILE reference sequence file (force -S) [null] 使用序列fasta文件作为header的输入
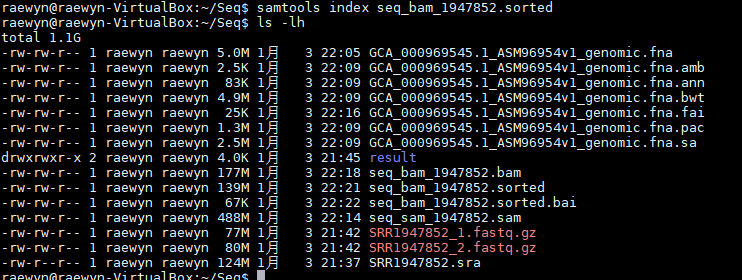
3、为bam文件排序,sort只能为bam文件排序,而不能为sam
samtools sort seq_bam_1947852.bam -o seq_bam_1947852.sorted

4、为排序的bam文件建立索引. *.bai文件
samtools index seq_bam_1947852.sorted
5、生成bcf文件
samtools mpileup -gf GCA_000969545.1_ASM96954v1_genomic.fna seq_bam_1947852.sorted > seq_1947852.bcf

语法详解:
samtools mpileup 生成bcf文件,bcf文件是二进制文件
-g, --BCF 生成bcf格式文件;
-f , --fasta-ref FILE参考序列(fasta format)的索引文件名;
Step 6 变异检测
软件:bcftools
步骤:
1、变异检测
bcftools call -vm seq_1947852.bcf -o seq_1947852.variants.bcf
2、将bcf文件转换为vcf文件
bcftools view -v snps,indels seq_1947852.variants.bcf > seq_1947852.snps.vcf
3、变异位点的过滤
bcftools filter -o seq_1947852.snps.filtered.vcf -i 'QUAL>20 && DP>5' seq_1947852.snps.vcf
简单过滤:QUAL小于等于20,DP值小于等于5的位点
-i – include 只保留满足该条件的位点
Step 7 变异基因注释
软件:annovar
1、生成annovar输入文件
convert2annovar.pl -format vcf4 seq_1947852.snps.filtered.vcf > seq_1947852.snps.avinput
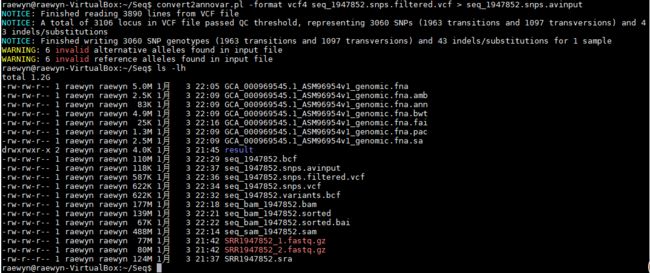
2、注释变异基因位点
annotate_variation.pl --geneanno --dbtype refGene --buildver 1947852-genome seq_1947852.snps.avinput ~/Biosoft/annovar/humandb/
--geneanno: 采用gene-based annotation注释模式;
--dbtype :数据库为refGene;还有knowGene,ensGene等
--buildver:为参考基因组版本,需特别注意
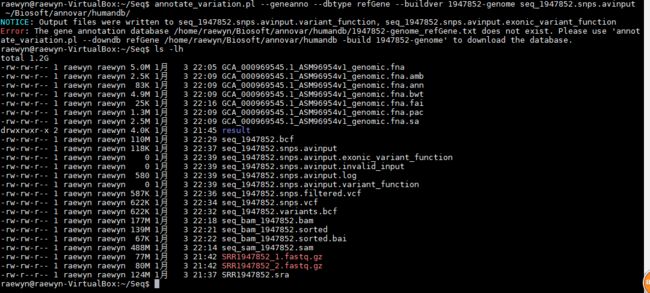
没有结果,需要自定义数据库(后面有讲解)
3、最后为数据库所在目录,生成avinput.variant_function和avinput.exonic_variant_function后缀的两个结果文件
Annovar结果解析
①avinput.variant_function文件
包括所有突变信息:
第一列:variant effects, 变异分类,如intergenic, intronic, nonsynonymous SNP, frameshift deletion, large-scale duplication等
第二列:基因名或Symbol
其余列:与avinput输入文件相同:染色体、start、end、ref_nt、obs_nt等
②avinput.exonic_variant_function文件
包括外显子突变信息
第一列:该变异在input文件的行号
第二列:对编码基因的影响,frameshift, synonymous or nonsynonymous等
第三列:被影响的基因或转录本,包括染色体、基因起始、核苷酸和氨基酸突变的
情况
其余列:同输入文件其余列:与avinput输入文件相同:染色体、start、end、ref_nt、obs_nt等
自定义数据库
1、下载基因组gff文件
wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/969/545/GCA_000969545.1_ASM96954v1/GCA_000969545.1_ASM96954v1_genomic.gff.gz
gunzip GCA_000969545.1_ASM96954v1_genomic.gff.gz
2:、gff文件转为GenePred文件
wget http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/gff3ToGenePred
chmod 777 gff3ToGenePred
./gff3ToGenePred -useName GCA_000969545.1_ASM96954v1_genomic.gff 1947852-genome_refGene.txt
3、为建立每个基因与编码序列对应文件
retrieve_seq_from_fasta.pl -format refGene -seqfile GCA_000969545.1_ASM96954v1_genomic.fna -outfile 1947852-genome_refGeneMrna.fa 1947852-genome_refGene.txt
5、拷贝数据库文件到annovar安装目录humandb文件夹下
cp 1947852-genome_refGene* ~/Biosoft/annovar/humandb/