首先友情宣传生信技能树
- 全国巡讲:R基础,Linux基础和RNA-seq实战演练 : 预告:12月28-30长沙站
- 广州珠江新城GEO数据挖掘滚动开班
题目来源:http://www.bio-info-trainee.com/3750.html
作业1
请根据R包
org.Hs.eg.db找到下面ensembl 基因ID 对应的基因名(symbol)ENSG00000000003.13
ENSG00000000005.5
ENSG00000000419.11
ENSG00000000457.12
ENSG00000000460.15
ENSG00000000938.11提示:
library(org.Hs.eg.db)
g2s=toTable(org.Hs.egSYMBOL)
g2e=toTable(org.Hs.egENSEMBL)
BiocManager::install("org.Hs.eg.db")
library(org.Hs.eg.db)
id <- read.table("T1_ID.txt",col.names = "ensembl_id_all")
library(stringr)
id$ensembl_id <- str_split(id$ensembl_id_all,"[.]",simplify = T)[,1]
g2s <- toTable(org.Hs.egSYMBOL)
g2e <- toTable(org.Hs.egENSEMBL)
id <- merge(id,g2e,by="ensembl_id",all.x = T)
id <- merge(id,g2s,by="gene_id",all.x = T)
id <- id[order(id$ensembl_id_all),]
for (i in 1:nrow(id)) {
print(paste(id$ensembl_id_all[i],'->',id$symbol[i]))
}
[1] "ENSG00000000003.13 -> TSPAN6"
[1] "ENSG00000000005.5 -> TNMD"
[1] "ENSG00000000419.11 -> DPM1"
[1] "ENSG00000000457.12 -> SCYL3"
[1] "ENSG00000000460.15 -> C1orf112"
[1] "ENSG00000000938.11 -> FGR"
作业 2
根据R包
hgu133a.db找到下面探针对应的基因名(symbol)1053_at
117_at
121_at
1255_g_at
1316_at
1320_at
1405_i_at
1431_at
1438_at
1487_at
1494_f_at
1598_g_at
160020_at
1729_at
177_at提示:
library(hgu133a.db)
ids=toTable(hgu133aSYMBOL)
head(ids)
BiocManager::install("hgu133a.db")
library(hgu133a.db)
ids=toTable(hgu133aSYMBOL)
head(ids)
Pid <- read.table("T2-probe_id.txt",col.names = "probe_id")
Pid <- merge(Pid,ids,by = "probe_id",all.x = T)
Pid
# probe_id symbol
#1 1053_at RFC2
#2 117_at HSPA6
#3 121_at PAX8
#4 1255_g_at GUCA1A
#5 1316_at THRA
#6 1320_at PTPN21
#7 1405_i_at CCL5
#8 1431_at CYP2E1
#9 1438_at EPHB3
#10 1487_at ESRRA
#11 1494_f_at CYP2A6
#12 1598_g_at GAS6
#13 160020_at MMP14
#14 1729_at TRADD
#15 177_at PLD1
作业 3
找到R包
CLL内置的数据集的表达矩阵里面的TP53基因的表达量,并且绘制在progres.-stable分组的boxplot图提示:
suppressPackageStartupMessages(library(CLL))
data(sCLLex)
sCLLex
exprSet=exprs(sCLLex)
library(hgu95av2.db)想想如何通过
ggpubr进行美化。

> suppressPackageStartupMessages(library(CLL))
> data(sCLLex)
> sCLLex
ExpressionSet (storageMode: lockedEnvironment)
assayData: 12625 features, 22 samples
element names: exprs
protocolData: none
phenoData
sampleNames: CLL11.CEL CLL12.CEL ... CLL9.CEL (22 total)
varLabels: SampleID Disease
varMetadata: labelDescription
featureData: none
experimentData: use 'experimentData(object)'
Annotation: hgu95av2
> exprSet=exprs(sCLLex)
> pd <- pData(sCLLex)
> library(hgu95av2.db)
> ids <- toTable(hgu95av2SYMBOL)
> tp53choose <- ids[,2]=="TP53"
> TP53_probe_id<- ids[tp53choose,][1]
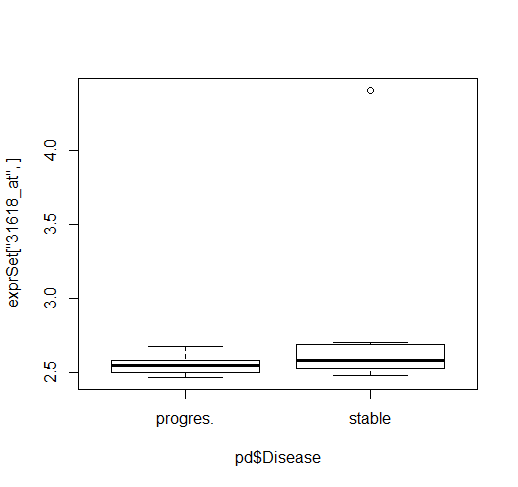
> TP53_probe_id
probe_id
966 1939_at
997 1974_s_at
1420 31618_at
#对应TP53基因有3个探针,分别绘制boxplot
>boxplot(exprSet["1939_at",]~pd$Disease)
>boxplot(exprSet["1974_s_at",]~pd$Disease)
>boxplot(exprSet["31618_at",]~pd$Disease)


作业 4
找到BRCA1基因在TCGA数据库的乳腺癌数据集(Breast Invasive Carcinoma (TCGA, PanCancer Atlas))的表达情况
提示:使用http://www.cbioportal.org/index.do 定位数据集:http://www.cbioportal.org/datasets
作业 5
找到TP53基因在TCGA数据库的乳腺癌数据集的表达量分组看其是否影响生存
提示使用:http://www.oncolnc.org/
作业6
下载数据集GSE17215的表达矩阵并且提取下面的基因画热图
ACTR3B ANLN BAG1 BCL2 BIRC5 BLVRA CCNB1 CCNE1 CDC20 CDC6 CDCA1 CDH3 CENPF CEP55 CXXC5 EGFR ERBB2 ESR1 EXO1 >FGFR4 FOXA1 FOXC1 GPR160 GRB7 KIF2C KNTC2 KRT14 KRT17 KRT5 MAPT MDM2 MELK MIA MKI67 MLPH MMP11 MYBL2 MYC NAT1 >ORC6L PGR PHGDH PTTG1 RRM2 SFRP1 SLC39A6 TMEM45B TYMS UBE2C UBE2T
提示:根据基因名拿到探针ID,缩小表达矩阵绘制热图,没有检查到的基因直接忽略即可。
rm(list = ls())
if (!require(AnnoProbe)) {
devtools::install_local("D:/biosoft/R/github/AnnoProbe.zip")
}
library(AnnoProbe)
gset=AnnoProbe::geoChina('GSE17215')
eSet=gset[[1]]
probe_expr <- exprs(eSet)
group_list <- c(rep('paclitaxel',3),rep('salinomycin',3))
library(hgu133a.db)
ids <- toTable(hgu133aSYMBOL)
p2g <- read.table("T6.txt",sep = ' ',col.names = "symbol")
p2g <- merge(p2g,ids,by = "symbol")
tmp <- p2g
p2g$mean <- rowMeans(probe_expr[tmp$probe_id,])
p2g <- p2g[order(p2g$symbol),]
p2g <- p2g[!duplicated(p2g$symbol),]
genes_heatmap <- probe_expr[p2g$probe_id,]
genes_heatmap <- t(scale(t(genes_heatmap)))
rownames(genes_heatmap) <- p2g$symbol
annotation_col <- data.frame(groups = group_list)
rownames(annotation_col) <- colnames(genes_heatmap)
pheatmap::pheatmap(genes_heatmap,filename = 'T6_heatmap.png',
annotation_col = annotation_col
)

作业7
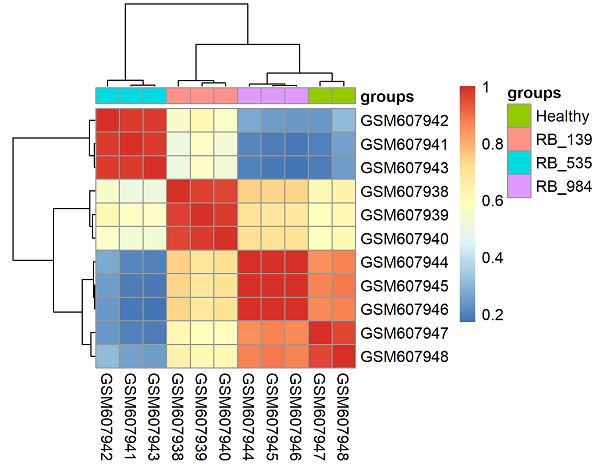
下载数据集GSE24673的表达矩阵计算样本的相关性并且绘制热图,需要标记上样本分组信息
rm(list=ls())
options(stringsAsFactors = F)
gset <- AnnoProbe::geoChina('GSE24673')
eSet <- gset[[1]]
exprSet <- exprs(eSet)
group_list <- read.table('Q7.txt')
rownames(group_list) <- group_list[,1]
group_list <- group_list[,-1]
gl <- data.frame(groups = group_list)
rownames(gl) <- colnames(exprSet)
names(group_list) <- 'groups'
dim(exprSet)
exprSet <- exprSet[apply(exprSet,1,function(x) sum(x>0)>5),]
if (!require(edgeR)) {
BiocManager::install('edgeR')
}
exprSet <- log(edgeR::cpm(exprSet)+1)
exprSet <- exprSet[names(sort(apply(exprSet,1,mad),decreasing = T)[1:500]),]
M <- cor(log2(exprSet+1))
pheatmap::pheatmap(M,annotation_col = gl,filename = 'Q7_heatmap.png')
作业8
找到 GPL6244 platform of Affymetrix Human Gene 1.0 ST Array 对应的R的bioconductor注释包,并且安装它!
options()$repos options()$BioC_mirror options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/") options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/")) BiocManager::install("请输入自己找到的R包",ask = F,update = F) options()$repos options()$BioC_mirror
参考:http://www.bio-info-trainee.com/1399.html

if (!require(hugene10sttranscriptcluster.db)) {
BiocManager::install("hugene10sttranscriptcluster.db")
}
library(hugene10sttranscriptcluster.db)
作业9
下载数据集GSE42872的表达矩阵,并且分别挑选出所有样本的(平均表达量/sd/mad/)最大的探针,并且找到它们对应的基因。
rm(list=ls())
options(stringsAsFactors = F)
library(AnnoProbe)
library(Biobase)
gset <- geoChina('GSE42872')
eSet <- gset[[1]]
exprSet <- exprs(eSet)
library(hugene10sttranscriptcluster.db)
IDs <- toTable(hugene10sttranscriptclusterSYMBOL)
IDs_means_max <- sort(apply(exprSet,1,mean),decreasing = T)[1]
names(IDs_means_max)
#[1] "7978905"
IDs_sd_max <- sort(apply(exprSet,1,sd),decreasing = T)[1]
names(IDs_sd_max)
#[1] "8133876"
IDs_mad_max <- sort(apply(exprSet,1,mad),decreasing = T)[1]
names(IDs_mad_max)
#[1] "8133876"
table(IDs$probe_id == names(IDs_means_max))
#FALSE
#19814
table(IDs$probe_id == names(IDs_sd_max))
#FALSE TRUE
#19813 1
IDs[IDs$probe_id == names(IDs_sd_max),]
# probe_id symbol
#16463 8133876 CD36
table(IDs$probe_id == IDs_mad_max)
#FALSE TRUE
#19813 1
#IDs[IDs$probe_id == names(IDs_mad_max),]
# probe_id symbol
#16463 8133876 CD36
IDs_means <- sort(apply(exprSet,1,mean),decreasing = T)
i <- 1
while (!(names(IDs_means)[i] %in% IDs$probe_id)) { i <- i+1}
i
#[1] 6
#平均表达量从高到低排序,前5个探针均没有对应的基因symbol
names(IDs_means)[i]
#[1] "7953385"
IDs[IDs$probe_id==names(IDs_means)[i],]
# probe_id symbol
#4004 7953385 GAPDH
>
作业10
下载数据集GSE42872的表达矩阵,并且根据分组使用limma做差异分析,得到差异结果矩阵
参考:limma包的使用技巧
rm(list=ls())
options(stringsAsFactors = F)
library(AnnoProbe)
library(Biobase)
gset <- geoChina('GSE42872')
eSet <- gset[[1]]
exprSet <- exprs(eSet)
pd <- pData(eSet)
group_list <- c(rep('Control',3),rep('Vemurafenib',3))
group_list <- c(rep('Control',3),rep('Vemurafenib',3))
group_list <- data.frame(group_list)
rownames(group_list) <- colnames(exprSet)
library(limma)
design <- model.matrix(~0+as.factor(group_list$group_list))
colnames(design)<-c('Control','Vemurafenib')
fit <- lmFit(exprSet,design)
contrast.matrix <- makeContrasts(Control-Vemurafenib,levels = design)
fit2 <- contrasts.fit(fit,contrast.matrix)
fit2 <- eBayes(fit2)
DEG <- topTable(fit2,coef = 1,n=Inf)
nrDEG <- na.omit(DEG)
#Visualization
p_thred <- 0.05
logFC_thred <- 2
p4draw <- function(DEG,p_thred,logFC_thred) {
pp <- data.frame(symbols=rownames(DEG),
logFC=DEG$logFC,
P.Value=DEG$P.Value)
pp$groups = ifelse(pp$P.Value > p_thred, "stable",
ifelse(pp$logFC > logFC_thred, "up",
ifelse(pp$logFC < -logFC_thred, "down",
"stable")))
return(pp)
}
pp <- p4draw(nrDEG,p_thred,logFC_thred)
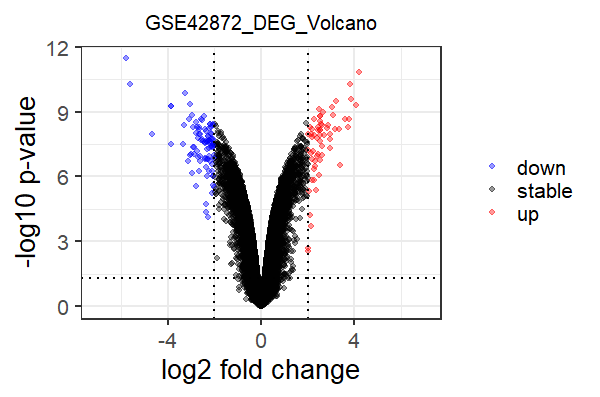
table(pp$groups)
# down stable up
# 92 33144 61
library(ggplot2)
drawVolcano <- function(pp){
g = ggplot(data = pp, aes(x = logFC, y = -log10(P.Value), color = groups)) +
geom_point(alpha = 0.4, size = 1.75) +
geom_vline(xintercept = c(-2,2),lty=3,lwd=0.8) +
geom_hline(yintercept=-log10(0.05),lty=3,lwd=0.8) +
xlim(c(-7,7)) +
theme_set(theme_set(theme_bw(base_size = 20))) +
labs(x="log2 fold change",y="-log10 p-value",title="GSE42872_DEG_Volcano") +
theme(plot.title = element_text(size = 15,hjust = 0.5)) +
scale_colour_manual(values = c("blue","black", "red")) +
theme(legend.title = element_blank())
return(g)
}
drawVolcano(pp)