scATAC分析神器ArchR初探-简介(1)
scATAC分析神器ArchR初探-ArchR进行doublet处理(2)
scATAC分析神器ArchR初探-创建ArchRProject(3)
scATAC分析神器ArchR初探-使用ArchR降维(4)
scATAC分析神器ArchR初探--使用ArchR进行聚类(5)
scATAC分析神器ArchR初探-单细胞嵌入(6)
scATAC分析神器ArchR初探-使用ArchR计算基因活性值和标记基因(7)
scATAC分析神器ArchR初探-scRNA-seq确定细胞类型(8)
scATAC分析神器ArchR初探-ArchR中的伪批次重复处理(9)
scATAC分析神器ArchR初探-使用ArchR-peak-calling(10)
scATAC分析神器ArchR初探-使用ArchR识别标记峰(11)
scATAC分析神器ArchR初探-使用ArchR进行主题和功能丰富(12)
scATAC分析神器ArchR初探-利用ArchR丰富ChromVAR偏差(13)
scATAC分析神器ArchR初探-使用ArchR进行足迹(14)
scATAC分析神器ArchR初探-使用ArchR进行整合分析(15)
scATAC分析神器ArchR初探-使用ArchR进行轨迹分析(16)
10-使用ArchR-peak-calling
peak-calling是ATAC-seq数据分析中最基本的过程之一。因为每单元scATAC-seq数据本质上是二进制的(可访问或不可访问),所以我们不能在单个单元上peak-calling。因此,我们在上一章中定义了单元格组,通常是群集。此外,我们创建了伪大量复制品,以使我们能够评估峰调用的可重复性。
10.1迭代重叠峰合并过程
我们首先在2018年引入了迭代重叠峰合并的策略。其他峰值合并策略也遇到一些我们在下面概述的关键问题。
10.1.1固定宽度与可变宽度峰
我们使用501-bp的固定宽度峰,因为它们不需要归一化峰长,因此使下游计算更容易。此外,ATAC-seq中的绝大多数峰宽都小于501-bp。使用可变宽度的峰也很难合并来自多个样本的峰调用。通常,我们认为使用可变宽度峰带来的潜在收益并不超过成本。更广泛地说,大多数分析相对于使用的峰集或峰样式是稳定的。
下面,我们使用具有几个不同峰的几种细胞类型的相同玩具示例,来说明这些常用峰合并方法之间的差异。
10.1.2使用bedtools合并原始峰重叠
原始峰重叠包括将任何彼此重叠的峰合并为一个更大的峰。在此方案中,菊花链成为一个大问题,因为彼此不直接重叠的峰包含在同一较大的峰中,因为它们被共享的内部峰桥接。这种方法的另一个问题是,如果您要跟踪峰顶,则必须为每个新的合并峰选择一个新峰,或者跟踪适用于每个新峰的所有峰。 。通常,这种峰合并方法是使用bedtools merge命令实现的。
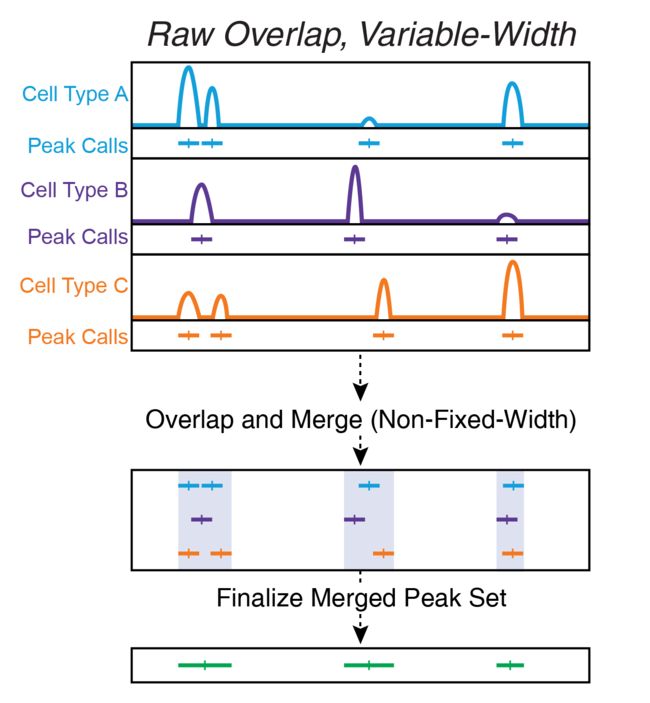
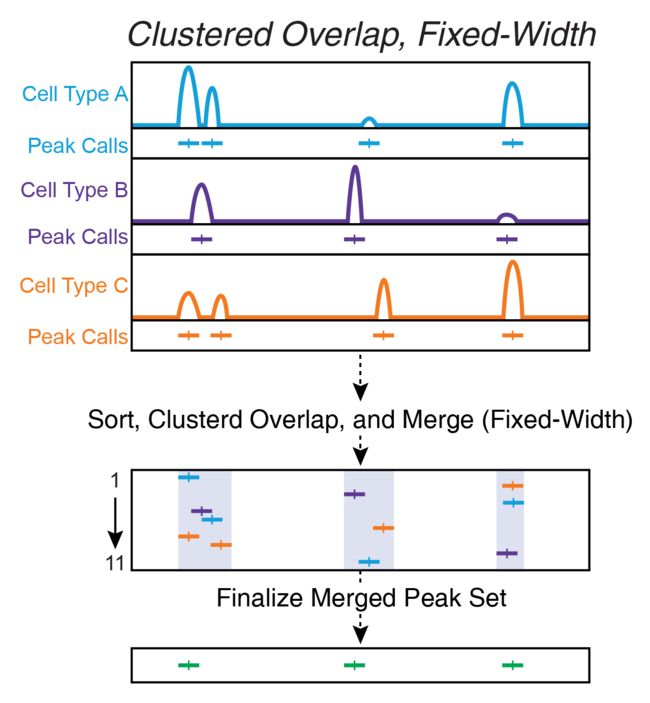
10.1.3使用bedtools群集的群集重叠
聚集的重叠峰聚集在一起,并选出一个获胜者。这通常是通过使用bedtools cluster命令并在每个群集中保留最重要的峰值来完成的。根据我们的经验,这最终会到达呼入峰,而错过附近的较小峰。
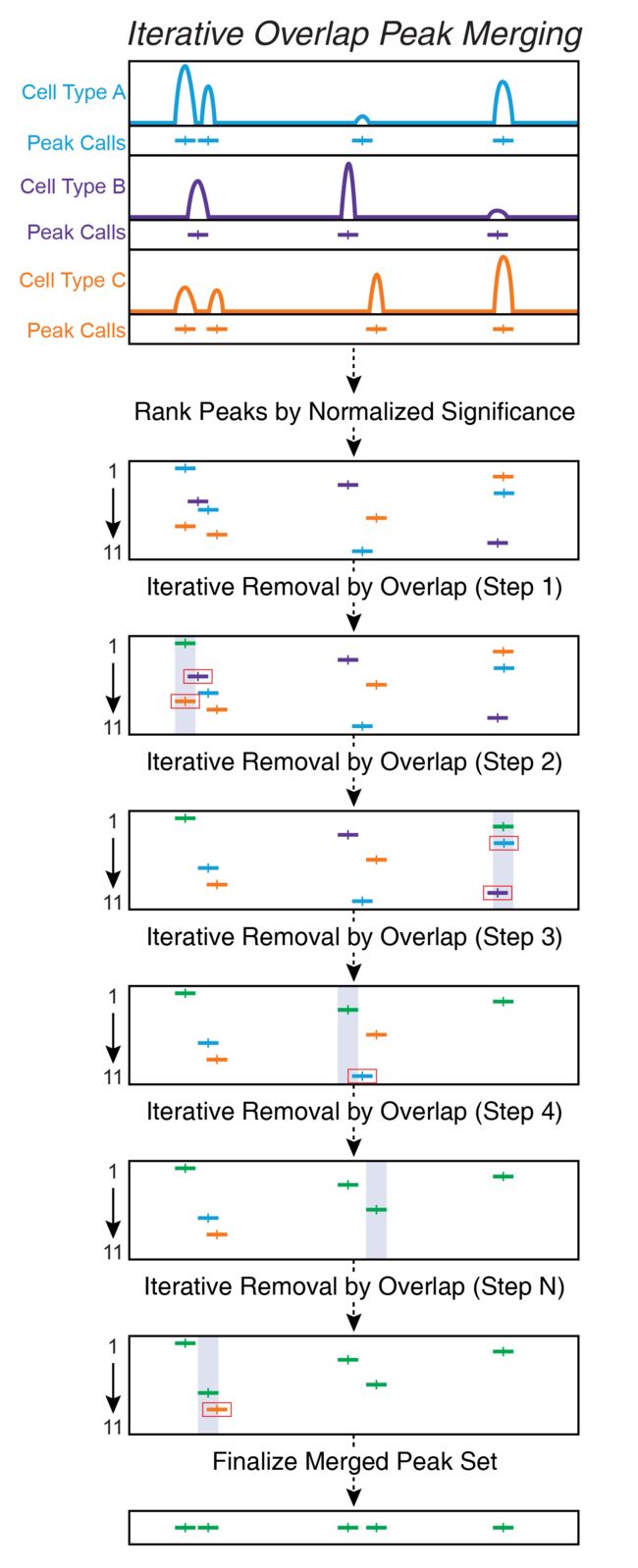
10.1.4 ArchR中的迭代重叠
迭代重叠删除可避免上述问题。峰首先按其重要性排序。保留最高有效峰,并将与最高有效峰直接重叠的任何峰从进一步分析中除去。然后,在剩余的峰中,重复此过程,直到不再存在任何峰为止。这样可以避免菊花链,并且仍然允许使用固定宽度的峰。
10.1.5高峰调用方式比较
比较所有这些方法产生的峰调用,可以直接看出最终峰集中的明显差异。我们认为,迭代重叠峰合并过程可产生具有最少警告的最佳峰集。

10.1.6那么这一切在ArchR中如何工作?
迭代重叠峰合并过程以分层方式执行,以最佳地保留特定于细胞类型的峰。
想象一下,您有3种细胞类型(A,B和C),每种细胞类型都有3个伪大量复制品。ArchR使用称为addReproduciblePeakSet()执行此迭代重叠峰合并过程。首先,ArchR会分别为每个伪批量复制调用峰。然后,ArchR将分析单个单元格类型的所有伪批量复制,一起执行迭代重叠移除的第一次迭代。重要的是要注意,ArchR对峰使用归一化的显着性度量来比较在不同样本中调用的峰的显着性。这是因为报告的MACS2显着性与测序深度成正比,因此峰的显着性在样品之间无法立即比较。在迭代重叠移除的第一次迭代之后,ArchR检查以查看伪批量重复中每个峰的重现性,并且仅保留通过该峰指示的阈值的峰。reproducibility参数。在此过程结束时,我们将为A,B和C这3种像元中的每一种都有一个合并的峰集。
然后,我们将重复此过程以合并A,B和C峰集。为此,我们将不同单元格类型的峰显着性重新归一化,并执行迭代重叠去除。最终结果是固定宽度峰的单个合并峰集。
10.1.7如果我不喜欢这种迭代的重叠峰合并过程,该怎么办?
迭代重叠峰合并过程由ArchR通过来实现,addReproduciblePeakSet()但您始终可以通过使用自己的峰集ArchRProj <- addPeakSet()。
10.2使用 Macs2调用峰
如上所述,我们使用该addReproduciblePeakSet()函数在ArchR中生成了可重现的峰集。默认情况下,ArchR尝试使用MACS2调用峰;但是,ArchR还实现了自己的本机峰值调用程序,当无法安装MACS2(例如,我们尚未在Windows上成功安装MACS2)时,可以使用该调用程序-下一节将介绍这种替代的峰值调用方法。
要使用MACS2调用峰,ArchR必须能够找到MACS2可执行文件。首先,ArchR查找您的PATH环境变量。如果失败,则ArchR尝试确定您是否已使用pip或来安装MACS2 pip3。如果这些都不成功,则ArchR放弃并提供错误消息。如果已安装MACS2,但ArchR找不到它,则应addReproduciblePeakSet()通过pathToMacs2参数提供该功能的路径。
pathToMacs2 <- findMacs2()
确定了通往MACS2的路径后,我们便可以创建带有MACS2的可重现合并峰集(约5-10分钟)。为了避免单元数很少的伪批量复制产生偏差,我们可以通过peaksPerCell参数为每个单元调用的峰数上限提供一个截止值。这样可以防止单元格很少的群集将大量低质量峰贡献给合并的峰集。还有许多其他参数可以调整addReproduciblePeakSet()-尝试?addReproduciblePeakSet获取更多信息。
每个ArchRProject对象只能包含一个峰集。因此,我们将输出分配给addReproduciblePeakSet()期望的ArchRProject。如果要尝试使用不同的峰集,则必须保存您的副本,ArchRProject从而也复制Arrow文件。尽管这确实占用了更多的磁盘存储空间,但是鉴于Arrow文件的结构以及在Arrow文件中存储峰矩阵信息,这是不可避免的。
projHeme4 <- addReproduciblePeakSet(
ArchRProj = projHeme4,
groupBy = "Clusters2",
pathToMacs2 = pathToMacs2
)
要检索此峰集作为GRanges对象,我们使用getPeakSet()函数。此峰集包含每个峰起源的组的注释。但是,这些注释并不固有地意味着仅在该组中调用给定的峰,而是带注释的组对该峰调用具有最高的归一化意义。
getPeakSet(projHeme4)
10.3使用 TileMatrix调用峰
如前所述,ArchR还实现了其自己的本机峰值调用程序。尽管我们已针对MACS2对该峰值调用者进行了基准测试,并注意到性能非常相似,但除非绝对必要,否则我们不建议您使用此本地峰值调用者。
ArchR本机峰值调用者在500 bp处调用峰TileMatrix,我们表明addReproduciblePeakSet()我们想通过peakMethod参数使用该峰值调用者。请注意,我们没有将输出存储到projHeme4对象中,因为我们不打算保持此峰值不变,并且此分析仅用于说明目的。存储到ArchRProject对象中将覆盖已经存储在中的先前可重现峰集projHeme4。
projHemeTmp <- addReproduciblePeakSet(
ArchRProj = projHeme4,
groupBy = "Clusters2",
peakMethod = "Tiles",
method = "p"
)
我们可以类似地检查此合并的峰集。
getPeakSet(projHemeTmp)
10.4添加峰矩阵
现在,我们可以projHeme4使用saveArchRProject()函数保存原始图像。这ArchRProject包含MACS2衍生的合并峰集。
saveArchRProject(ArchRProj = projHeme4, outputDirectory = "Save-ProjHeme4", load = FALSE)
要为下游分析做准备,我们可以创建一个新的ArchRProject名为projHeme5并添加新的基质含有成为我们新合并的波峰集合内插入计数。
projHeme5 <- addPeakMatrix(projHeme4)
现在我们可以看到已将一个新矩阵添加到projHeme5“ PeakMatrix”中。这是另一个类似于GeneScoreMatrix和的保留名称矩阵TileMatrix。如前所述,每个ArchRProject对象只能有一个峰集和一个峰集PeakMatrix。当然,您可以创建数量不限的不同名称的自定义特征矩阵,但是PeakMatrix保留给从存储在中的峰集派生的插入计数矩阵ArchRProject。
getAvailableMatrices(projHeme5)
参考材料:
https://www.archrproject.com/