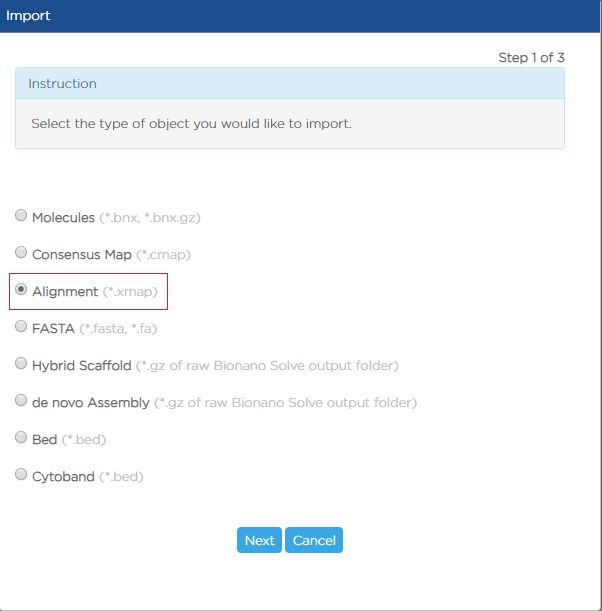
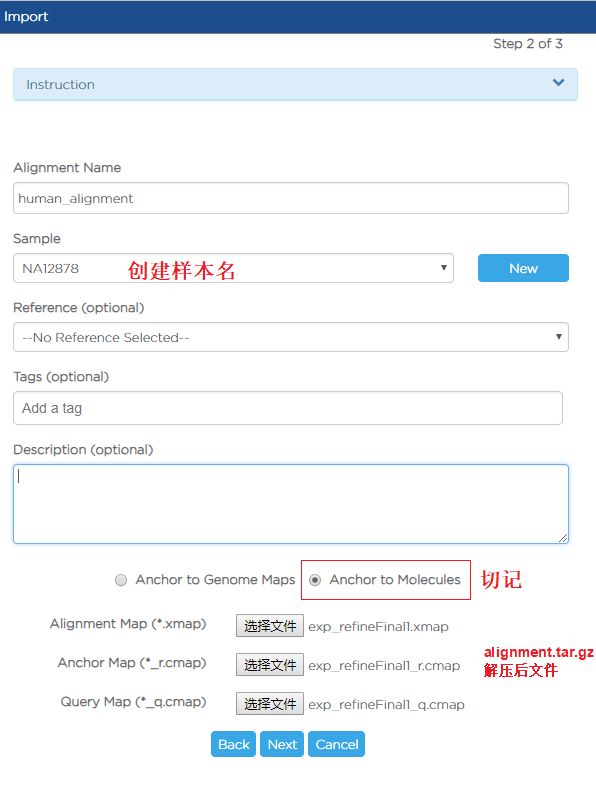
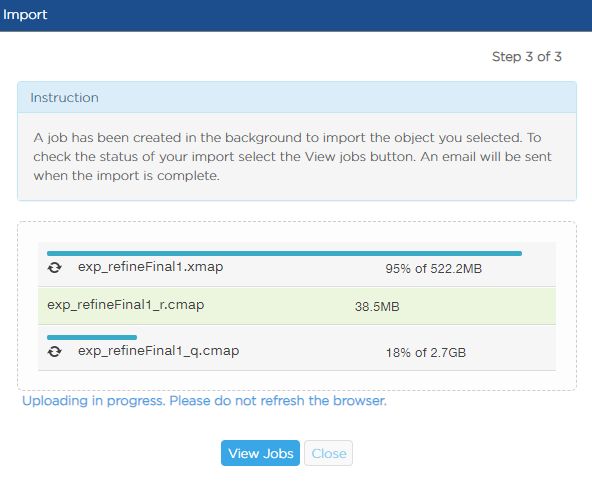
- C++中explicit类型转换运算符
水瓶丫头站住
C++关键字c++开发语言
在C++中,explicit类型转换运算符是用于防止隐式类型转换的关键特性。它主要应用于类的类型转换运算符(如operatortype()),确保类型转换必须通过显式调用来触发,从而提高代码安全性和可读性。以下是详细解析:核心概念基本语法classMyClass{public:explicitoperatorint()const{returnvalue;}private:intvalue;};ex
- Mysql-InnoDB索引:普通索引、主键索引、唯一索引、组合索引
豪大大ya
mysql数据库java
InnoDB和MyISAM的区别事务方面InnoDB支持事务,MyISAM不支持事务。这是Mysql将默认存储引擎从MyISAM变成InnoDB的重要原因之一外键方面InnoDB支持外键,而MyISAM不支持。对一个包含外键的InnoDB表转为MyISAM会失败索引层面InnoDB是聚集(聚簇)索引,MyISAM是非聚集(非聚簇)索引。MyISAM支持FULLTEXT类型的全文索引。InnoDB不
- 谈为什么KLA和Camtech公司为什么可以做到,半导体那边,晶圆,键合可以做到不管哪款新产品进来。编程2小时,上线后准确率可以直接做到99.9%、
*Major*
机器视觉
谈为什么KLA和Camtech公司为什么可以做到,半导体那边,晶圆,键合可以做到不管哪款新产品进来。编程2小时,上线后准确率可以直接做到99.9%、这么里面的AI原理没什么,还是这些公司把AI技术层面用出花了,一是他们有公司可能比较成立时间长,数据丰富。二是像AI深度学习网络冻结,或者自适应调参,都是一些AI技巧,他们用的比较好。三什么跨层特征解耦,实现的基础是他们对半导体理解比较深刻KLA和Ca
- python+flask实现360全景图和stl等多种格式模型浏览
mosquito_lover1
python
1.安装依赖pipinstallflask2.创建Flask应用创建一个基本的Flask应用,并设置路由来处理不同的文件类型。fromflaskimportFlask,render_template,send_from_directoryapp=Flask(__name__)#设置静态文件路径app.static_folder='static'@app.route('/')defindex():r
- 【QT教程】QT6硬件数据库编程 QT硬件数据库
QT性能优化QT原理源码QT界面美化
qtqt6.3qt5c++QT教程
QT6硬件数据库编程使用AI技术辅助生成QT界面美化视频课程QT性能优化视频课程QT原理与源码分析视频课程QTQMLC++扩展开发视频课程免费QT视频课程您可以看免费1000+个QT技术视频免费QT视频课程QT统计图和QT数据可视化视频免费看免费QT视频课程QT性能优化视频免费看免费QT视频课程QT界面美化视频免费看1QT6硬件数据库编程基础1.1QT6数据库引擎概述1.1.1QT6数据库引擎概述
- 深入解析:构建高效单页应用(SPA)的最佳实践与示例
布兰妮甜
#Vue单页应用SPAVue.js前端
文章目录前言一、单页应用(SPA)的介绍二、单页应用(SPA)的优势三、构建单页应用(SPA)的基本步骤四、使用Vue.js构建一个简易的单页应用(SPA):任务管理器结语前言随着互联网技术的发展,用户对于网页应用的交互性和响应速度提出了更高的要求。传统的多页面应用(MPA)在每次用户交互时需要重新加载整个页面,这不仅增加了服务器的负担,也降低了用户体验。而单页应用(SinglePageAppli
- Java短信模块开发-腾讯云短信服务
Hbb123654
腾讯云java
1、提前配置1、已有腾讯云账号和服务器2、开通短信服务,创建签名和模板并通过审核,如:国内短信快速入门3、需要先购买国内短信套餐包。4、在访问管理控制台>API密钥管理页面获取SecretID和SecretKey。5、安装最新版本的Maven依赖com.tencentcloudapitencentcloud-sdk-java3.1.10002、Java代码逻辑实例1、短信工具类方法/***Tenc
- 是德科技N9020A使用领域介绍
圣格特刘工
嵌入式硬件网络可用性测试集成测试人工智能
是德科技(Keysight)N9020A频谱分析仪是一款高性能信号分析仪,适用于多个领域,主要包括以下应用场景:1.无线通信测试5G/4G/LTE/Wi-Fi测试:用于评估通信系统的射频性能,分析信号质量、调制精度等。射频器件测试:测量功率、带宽、谐波失真等指标,确保射频元器件符合标准。2.雷达与国防雷达信号分析:测试雷达信号的频谱特性、瞬时带宽、脉冲宽度等参数。电子战(EW)测试:分析复杂电磁环
- 30、map 和 unordered_map的区别和实现机制【高频】
桃酥403
桃酥的学习笔记(C++篇)哈希算法算法
底层结构map底层是红黑树结构,而unordered_map底层是哈希结构;有序性但是红黑树其实是一种二叉搜索树,插入删除时会自动排序hash因为是把数据映射到数组上的,而且存在哈希冲突,所以不能保证有序存储所以有序存储使用map(红黑树的中序遍历,就能把储存的数据从小到大把数据按序展现出来)查找为了查找,红黑树需要依次比较关键码,时间复杂度为logn,还要加上平衡节点旋转的时间虽然说哈希表的内存
- Python爬虫教程:如何通过接口批量下载视频封面(FFmpeg技术实现)
Python爬虫项目
python爬虫开发语言数据库数据分析scrapyselenium
引言随着在线视频平台的蓬勃发展,视频封面作为视频内容的预览图,一直以来都是观众对视频的第一印象。在爬取视频资源时,很多开发者和研究者往往只关注视频本身,而忽略了视频封面。实际上,视频封面不仅能提供重要的信息(例如视频标题、主题或情感等),而且它们也能作为数据集中的重要属性,用于视频分类、推荐系统等应用。在这篇博客中,我们将深入探讨如何使用Python通过接口批量下载视频封面,利用FFmpeg等技术
- AI 大模型应用数据中心的数据分析架构
AI天才研究院
计算AI大模型企业级应用开发实战DeepSeekR1&大数据AI人工智能大模型javapythonjavascriptkotlingolang架构人工智能大厂程序员硅基计算碳基计算认知计算生物计算深度学习神经网络大数据AIGCAGILLM系统架构设计软件哲学Agent程序员实现财富自由
《AI大模型应用数据中心的数据分析架构》关键词:数据中心、AI大模型、数据分析、架构设计、应用实践摘要:本文深入探讨了AI大模型在数据中心数据分析架构中的应用,从数据中心背景、AI大模型架构与技术、数据处理与分析技术、AI大模型应用与实践等多个方面,全面解析了AI大模型如何助力数据中心实现高效数据分析和智能处理,为读者提供了系统的理论指导和实际案例分析。第一部分:数据中心背景与AI大模型概述第1章
- 审核通过≠报名成功!提醒下报了名的软考考生,这件事别忘了做!
公众号-希赛网
学习方法职场和发展
截止至3月13日18:00,江苏、贵州、山西、大连、安徽、福建、澳门、甘肃、新疆、兵团、四川、浙江、辽宁、吉林、宁波等考区均已开通报名入口。已经报名和准备报名的考生,小希提醒大家,审核通过≠报名成功!一、审核通过≠报名成功虽然软考对考生的学历、专业、工作年限等没有要求,但是部分考区还是会对考生上传的材料、填写的报考信息等进行审核,以确认证件照片满足要求并符合报考属地化管理原则。报名系统并不会以任何
- C# AOT生成的hellowwordEXE运行占用多少内存1-5MB?
专注VB编程开发20年
c#策略模式开发语言
C#使用AOT(Ahead-Of-Time,提前编译)生成的"Hello,World!"可执行文件在运行时占用的内存会受到多种因素的影响,以下是详细分析:影响内存占用的因素操作系统:不同的操作系统(如Windows、Linux、macOS)对进程的内存管理机制不同,会导致内存占用有所差异。运行环境:包括系统中已运行的其他程序、系统的内存管理策略等。编译器和运行时配置:不同版本的.NETSDK以及编
- C# HashTable、HashSet、Dictionary
有诗亦有远方
C#Hash
哈希一、HashTable1.什么是哈希表2.哈希表的Key&Value(1)添加数据(2)“键值对”均是object类型(3)必须有Key键,且Key键不能重复。(4)乱序读取数据3.基本操作二、HashSet1.特点2.HashSet常用扩展方法3.HashSet与Linq操作三、Dictionary四、HashTable和Dictionary的区别一、HashTable哈希表(HashTab
- 简单了解WIndow和Linux的路径含义
alive903
Linuxlinuxwindows
目录1>路径概念2>绝对路径2.1>window绝对路径2.2>Linux绝对路径3>相对路径3.1>window相对路径3.2>Linux相对路径很高兴你能看到这篇文章,同时我的语雀文档也更新了许多嵌入式系列的学习笔记希望能帮到你:https://www.yuque.com/alive-m4b9n1>路径概念路径是用来描述一个文件或目录在文件系统中的位置的方式。路径可以是文件系统中的唯一标识符,
- 嵌入式面试真题——Linux内核空间与用户空间
70000cc
嵌入式面试真题linuxc语言嵌入式硬件面试单片机
本文以32位系统为例介绍内核空间(kernelspace)和用户空间(userspace)。对32位操作系统而言,它的寻址空间(虚拟地址空间,或叫线性地址空间)为4G(2的32次方)。也就是说一个进程的最大地址空间为4G。操作系统的核心是内核(kernel),它独立于普通的应用程序,可以访问受保护的内存空间,也有访问底层硬件设备的所有权限。为了保证内核的安全,现在的操作系统一般都强制用户进程不能直
- MyBatis底层原理深度解析:动态代理与注解如何实现ORM映射
rider189
java开发语言mybatis
一、引言MyBatis作为一款优秀的ORM框架,其核心设计思想是通过动态代理和注解将接口方法与SQL操作解耦。开发者只需定义Mapper接口并添加注解,便能实现数据库操作,这背后隐藏着精妙的动态代理机制与源码设计。本文将从源码层解析MyBatis如何实现这一过程。二、动态代理机制:从接口到实现类关键点:MyBatis通过JDK动态代理为Mapper接口生成代理对象,拦截所有方法调用,将其路由到SQ
- 树莓科技集团董事长:第五代产业园运营模式的深度剖析与展望
树莓集团
科技人工智能百度物联网大数据
第五代产业园运营模式,以创新为核心驱动,强调数字化、网络化和资源整合。树莓科技集团在这一领域具有代表性,其运营模式值得深入剖析。核心特征数字化转型:第五代产业园高度重视数字化技术的应用,通过构建数字化平台,实现园区内企业、资源、信息的互联互通。并网化运营:树莓集团在全国28个省市布局产业园,形成网络化运营,促进资源共享和协同发展。全产业链整合:充分发挥全产业链资源整合优势,为入园企业提供全方位服务
- Python的struct模块
smilelance
Pythonpythonstructalignmentstringbufferexception
struct模块提供将二进制数据转换为结构化数据或相反的功能,它定义了以下函数和异常:exceptionstruct.errorstruct.pack(fmt,v1,v2,…)返回一个string,string由v1,v2…经过给出的格式fmt组成,参数的个数有和类型要和给出的格式一一对应struct.pack_into(fmt,buffer,offset,v1,v2,…)按照格式fmt将v1,v
- 【每日一题】93. 复原 IP 地址
聆听逝去的流
每日一题leetcode算法每日一题回溯
文章目录题目解题思路代码(C++)回溯总结题目题目链接:93.复原IP地址有效IP地址正好由四个整数(每个整数位于0到255之间组成,且不能含有前导0),整数之间用'.'分隔。例如:"0.1.2.201"和"192.168.1.1"是有效IP地址,但是"0.011.255.245"、"192.168.1.312"和"
[email protected]"是无效IP地址。给定一个只包含数字的字符串s,用以表示一个
- LeetCode-100题(Hot) 93. 复原 IP 地址 [Java实现]
IllTamer
LeetCodeleetcodejava
给定一个只包含数字的字符串,用以表示一个IP地址,返回所有可能从s获得的有效IP地址。你可以按任何顺序返回答案。有效IP地址正好由四个整数(每个整数位于0到255之间组成,且不能含有前导0),整数之间用'.'分隔。例如:"0.1.2.201"和"192.168.1.1"是有效IP地址,但是"0.011.255.245"、"192.168.1.312"和"
[email protected]"是无效IP地址。示例
- LeetCode详解C++版
纵深
算法算法数据结构c++
打算把LeetCode上面的题都实现一遍,每日两题LeetCode目录1.两数之和2.两数相加11.盛最多水的容器15.三数之和33.搜索旋转排序数组34.在排序数组中查找元素的第一个和最后一个位置35.搜索插入位置53.最大子数组和64.最小路径和70.爬楼梯74.搜索二维矩阵82.删除排序链表中的重复元素II88.合并两个有序数组153.寻找旋转排序数组中的最小值162.寻找峰值167.两数之
- python的一些基础知识学习
勇敢一点♂
python学习
列表(list)和元组(tuple)列表和元组,都是一个可以放置任意数据类型的有序集合,比如里面可以同时包含int和string类型都是有序的列表是动态的,长度大小不固定,可以随意地增加、删减或者改变元素。元组是静态的,长度大小固定,无法增加删减或者改变常规操作关于赋值,list可以很轻松的根据索引赋值,但是tuple不可以listA=[1,2,3,4]listA[3]=10print(listA
- 《Web 应用项目开发》课程心得体会:从理论到实战,开启 Web 开发新征程
m0_74824091
前端
在信息技术飞速发展的当下,Web应用已然渗透到生活的方方面面,从日常网购、社交娱乐,到在线办公、学习平台,无一不是Web应用的成果。怀着对互联网技术的热忱与憧憬,我踏入了《Web应用项目开发》这门课程,历经数月的沉浸式学习与实践,收获远超预期,犹如经历一场脱胎换骨的蜕变,以下便是我在这门课程中的全面心得体会。夯实基础:Web技术初相识课程伊始,仿若踏入一片未知的技术丛林,HTML、CSS和Java
- 3月14日复盘
四万二千
python人工智能
挑战AI全栈第四天!(终于双休了)容器python中默认有4种容器列表list字典dict集合set元组tuple一、Python列表(list)Python支持多种复合数据类型,可将不同值组合在一起。最常用的列表,是用方括号标注,逗号分隔的一组值。列表可以包含不同类型的元素,但一般情况下,各个元素的类型相同列表是一种可以存储任意个各种类型的序列容器列表内的数据有先后顺序关系列表是可变的容器1.列
- python内置函数 V
棠越精进
pythonpython开发语言
python内置函数VPython解释器内置了很多函数和类型,任何时候都能使用。V名称描述vars返回当前局部符号表的字典。vars()vars(object)返回模块、类、实例或任何其它具有__dict__属性的对象的__dict__属性。模块和实例这样的对象具有可更新的__dict__属性;但是,其它对象的__dict__属性可能会设为限制写入(例如,类会使用types.MappingProx
- C程序员驯服Common Lisp - 入门 - [语言探索]<转载>
acool555
lisp语言c编程documentationfortran
版权声明:转载时请以超链接形式标明文章原始出处和作者信息及本声明http://bigwhite.blogbus.com/logs/158733479.html毫无疑问,CommonLisp是一门庞大且复杂的语言,学习曲线并不平坦。对于一个从未接触过函数式语言、交互式语言以及动态类型语言的C程序员来说,学习CommonLisp显然是一个很大的挑战。也许有人会问:"C语言已经无所不能了,为何还要学习C
- 2025 年最值得收听的 AI 播客推荐!助你轻松掌握人工智能前沿动态!
真智AI
人工智能开发语言机器学习
如今,几乎每个人都被告知需要提升技能,而当前许多组织最看重的技能之一就是人工智能(AI)。学习AI相关技能通常涉及数学、统计学和机器学习,但除此之外,你还需要了解行业趋势、业内人士的观点以及各大公司的动态。然而,学习并不意味着时刻都要埋头苦读!有时候,你需要给大脑一个喘息的机会,同时依然能获取有价值的信息。而收听AI相关的播客,就是一个轻松高效的方式。以下是2025年你必须关注的AI播客!1.Th
- C# -Dictionary、HashTable、List、HashSet区别
※※冰馨※※
c#开发语言
在.Net模仿java的过程中,抛弃了HashMap,所以我们今天分析下Dictionary、HashTable、HashSet区别。处理碰撞,即碰撞到同一个Bucket槽上:Hashtable和Dictionary从数据结构上来说都属于Hashtable(哈希表),都是对关键字(键值)进行散列操作,将关键字散列到Hashtable的某一个槽位中去,不同的是处理碰撞的方法。散列函数有可能将不同的关
- 抗辐照CANFD芯片工艺解析:如何保障芯片的可靠性
国科安芯
科普嵌入式硬件安全威胁分析安全性测试
地面车规芯片容易受到大气中期效应的影响进而发生单粒子效应进而引起软错误,在航天领域这一问题又进一步细化为单粒子闩锁、单粒子翻转等问题。2024年,国内车厂也开始关注车规芯片的抗软错误问题,并做了一系列的实验。CANFD接口芯片作为在汽车电子学应用最多的芯片,在软错误防护上的考虑较为迫切。另一方面,在商业航天领域,为了降本增效,在通信接口的选择上越来越倾向于CANFD芯片。CANFD接口芯片不仅继承
- java观察者模式
3213213333332132
java设计模式游戏观察者模式
观察者模式——顾名思义,就是一个对象观察另一个对象,当被观察的对象发生变化时,观察者也会跟着变化。
在日常中,我们配java环境变量时,设置一个JAVAHOME变量,这就是被观察者,使用了JAVAHOME变量的对象都是观察者,一旦JAVAHOME的路径改动,其他的也会跟着改动。
这样的例子很多,我想用小时候玩的老鹰捉小鸡游戏来简单的描绘观察者模式。
老鹰会变成观察者,母鸡和小鸡是
- TFS RESTful API 模拟上传测试
ronin47
TFS RESTful API 模拟上传测试。
细节参看这里:https://github.com/alibaba/nginx-tfs/blob/master/TFS_RESTful_API.markdown
模拟POST上传一个图片:
curl --data-binary @/opt/tfs.png http
- PHP常用设计模式单例, 工厂, 观察者, 责任链, 装饰, 策略,适配,桥接模式
dcj3sjt126com
设计模式PHP
// 多态, 在JAVA中是这样用的, 其实在PHP当中可以自然消除, 因为参数是动态的, 你传什么过来都可以, 不限制类型, 直接调用类的方法
abstract class Tiger {
public abstract function climb();
}
class XTiger extends Tiger {
public function climb()
- hibernate
171815164
Hibernate
main,save
Configuration conf =new Configuration().configure();
SessionFactory sf=conf.buildSessionFactory();
Session sess=sf.openSession();
Transaction tx=sess.beginTransaction();
News a=new
- Ant实例分析
g21121
ant
下面是一个Ant构建文件的实例,通过这个实例我们可以很清楚的理顺构建一个项目的顺序及依赖关系,从而编写出更加合理的构建文件。
下面是build.xml的代码:
<?xml version="1
- [简单]工作记录_接口返回405原因
53873039oycg
工作
最近调接口时候一直报错,错误信息是:
responseCode:405
responseMsg:Method Not Allowed
接口请求方式Post.
- 关于java.lang.ClassNotFoundException 和 java.lang.NoClassDefFoundError 的区别
程序员是怎么炼成的
真正完成类的加载工作是通过调用 defineClass来实现的;
而启动类的加载过程是通过调用 loadClass来实现的;
就是类加载器分为加载和定义
protected Class<?> findClass(String name) throws ClassNotFoundExcept
- JDBC学习笔记-JDBC详细的操作流程
aijuans
jdbc
所有的JDBC应用程序都具有下面的基本流程: 1、加载数据库驱动并建立到数据库的连接。 2、执行SQL语句。 3、处理结果。 4、从数据库断开连接释放资源。
下面我们就来仔细看一看每一个步骤:
其实按照上面所说每个阶段都可得单独拿出来写成一个独立的类方法文件。共别的应用来调用。
1、加载数据库驱动并建立到数据库的连接:
Html代码
St
- rome创建rss
antonyup_2006
tomcatcmsxmlstrutsOpera
引用
1.RSS标准
RSS标准比较混乱,主要有以下3个系列
RSS 0.9x / 2.0 : RSS技术诞生于1999年的网景公司(Netscape),其发布了一个0.9版本的规范。2001年,RSS技术标准的发展工作被Userland Software公司的戴夫 温那(Dave Winer)所接手。陆续发布了0.9x的系列版本。当W3C小组发布RSS 1.0后,Dave W
- html表格和表单基础
百合不是茶
html表格表单meta锚点
第一次用html来写东西,感觉压力山大,每次看见别人发的都是比较牛逼的 再看看自己什么都还不会,
html是一种标记语言,其实很简单都是固定的格式
_----------------------------------------表格和表单
表格是html的重要组成部分,表格用在body里面的
主要用法如下;
<table>
&
- ibatis如何传入完整的sql语句
bijian1013
javasqlibatis
ibatis如何传入完整的sql语句?进一步说,String str ="select * from test_table",我想把str传入ibatis中执行,是传递整条sql语句。
解决办法:
<
- 精通Oracle10编程SQL(14)开发动态SQL
bijian1013
oracle数据库plsql
/*
*开发动态SQL
*/
--使用EXECUTE IMMEDIATE处理DDL操作
CREATE OR REPLACE PROCEDURE drop_table(table_name varchar2)
is
sql_statement varchar2(100);
begin
sql_statement:='DROP TABLE '||table_name;
- 【Linux命令】Linux工作中常用命令
bit1129
linux命令
不断的总结工作中常用的Linux命令
1.查看端口被哪个进程占用
通过这个命令可以得到占用8085端口的进程号,然后通过ps -ef|grep 进程号得到进程的详细信息
netstat -anp | grep 8085
察看进程ID对应的进程占用的端口号
netstat -anp | grep 进程ID
&
- 优秀网站和文档收集
白糖_
网站
集成 Flex, Spring, Hibernate 构建应用程序
性能测试工具-JMeter
Hmtl5-IOCN网站
Oracle精简版教程网站
鸟哥的linux私房菜
Jetty中文文档
50个jquery必备代码片段
swfobject.js检测flash版本号工具
- angular.extend
boyitech
AngularJSangular.extendAngularJS API
angular.extend 复制src对象中的属性去dst对象中. 支持多个src对象. 如果你不想改变一个对象,你可以把dst设为空对象{}: var object = angular.extend({}, object1, object2). 注意: angular.extend不支持递归复制. 使用方法: angular.extend(dst, src); 参数:
- java-谷歌面试题-设计方便提取中数的数据结构
bylijinnan
java
网上找了一下这道题的解答,但都是提供思路,没有提供具体实现。其中使用大小堆这个思路看似简单,但实现起来要考虑很多。
以下分别用排序数组和大小堆来实现。
使用大小堆:
import java.util.Arrays;
public class MedianInHeap {
/**
* 题目:设计方便提取中数的数据结构
* 设计一个数据结构,其中包含两个函数,1.插
- ajaxFileUpload 针对 ie jquery 1.7+不能使用问题修复版本
Chen.H
ajaxFileUploadie6ie7ie8ie9
jQuery.extend({
handleError: function( s, xhr, status, e ) {
// If a local callback was specified, fire it
if ( s.error ) {
s.error.call( s.context || s, xhr, status, e );
}
- [机器人制造原则]机器人的电池和存储器必须可以替换
comsci
制造
机器人的身体随时随地可能被外来力量所破坏,但是如果机器人的存储器和电池可以更换,那么这个机器人的思维和记忆力就可以保存下来,即使身体受到伤害,在把存储器取下来安装到一个新的身体上之后,原有的性格和能力都可以继续维持.....
另外,如果一
- Oracle Multitable INSERT 的用法
daizj
oracle
转载Oracle笔记-Multitable INSERT 的用法
http://blog.chinaunix.net/uid-8504518-id-3310531.html
一、Insert基础用法
语法:
Insert Into 表名 (字段1,字段2,字段3...)
Values (值1,
- 专访黑客历史学家George Dyson
datamachine
on
20世纪最具威力的两项发明——核弹和计算机出自同一时代、同一群年青人。可是,与大名鼎鼎的曼哈顿计划(第二次世界大战中美国原子弹研究计划)相 比,计算机的起源显得默默无闻。出身计算机世家的历史学家George Dyson在其新书《图灵大教堂》(Turing’s Cathedral)中讲述了阿兰·图灵、约翰·冯·诺依曼等一帮子天才小子创造计算机及预见计算机未来
- 小学6年级英语单词背诵第一课
dcj3sjt126com
englishword
always 总是
rice 水稻,米饭
before 在...之前
live 生活,居住
usual 通常的
early 早的
begin 开始
month 月份
year 年
last 最后的
east 东方的
high 高的
far 远的
window 窗户
world 世界
than 比...更
- 在线IT教育和在线IT高端教育
dcj3sjt126com
教育
codecademy
http://www.codecademy.com codeschool
https://www.codeschool.com teamtreehouse
http://teamtreehouse.com lynda
http://www.lynda.com/ Coursera
https://www.coursera.
- Struts2 xml校验框架所定义的校验文件
蕃薯耀
Struts2 xml校验Struts2 xml校验框架Struts2校验
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
蕃薯耀 2015年7月11日 15:54:59 星期六
http://fa
- mac下安装rar和unrar命令
hanqunfeng
mac
1.下载:http://www.rarlab.com/download.htm 选择
RAR 5.21 for Mac OS X 2.解压下载后的文件 tar -zxvf rarosx-5.2.1.tar 3.cd rar sudo install -c -o $USER unrar /bin #输入当前用户登录密码 sudo install -c -o $USER rar
- 三种将list转换为map的方法
jackyrong
list
在本文中,介绍三种将list转换为map的方法:
1) 传统方法
假设有某个类如下
class Movie {
private Integer rank;
private String description;
public Movie(Integer rank, String des
- 年轻程序员需要学习的5大经验
lampcy
工作PHP程序员
在过去的7年半时间里,我带过的软件实习生超过一打,也看到过数以百计的学生和毕业生的档案。我发现很多事情他们都需要学习。或许你会说,我说的不就是某种特定的技术、算法、数学,或者其他特定形式的知识吗?没错,这的确是需要学习的,但却并不是最重要的事情。他们需要学习的最重要的东西是“自我规范”。这些规范就是:尽可能地写出最简洁的代码;如果代码后期会因为改动而变得凌乱不堪就得重构;尽量删除没用的代码,并添加
- 评“女孩遭野蛮引产致终身不育 60万赔偿款1分未得”医腐深入骨髓
nannan408
先来看南方网的一则报道:
再正常不过的结婚、生子,对于29岁的郑畅来说,却是一个永远也无法实现的梦想。从2010年到2015年,从24岁到29岁,一张张新旧不一的诊断书记录了她病情的同时,也清晰地记下了她人生的悲哀。
粗暴手术让人发寒
2010年7月,在酒店做服务员的郑畅发现自己怀孕了,可男朋友却联系不上。在没有和家人商量的情况下,她决定堕胎。
12月5日,
- 使用jQuery为input输入框绑定回车键事件 VS 为a标签绑定click事件
Everyday都不同
jspinput回车键绑定clickenter
假设如题所示的事件为同一个,必须先把该js函数抽离出来,该函数定义了监听的处理:
function search() {
//监听函数略......
}
为input框绑定回车事件,当用户在文本框中输入搜索关键字时,按回车键,即可触发search():
//回车绑定
$(".search").keydown(fun
- EXT学习记录
tntxia
ext
1. 准备
(1) 官网:http://www.sencha.com/
里面有源代码和API文档下载。
EXT的域名已经从www.extjs.com改成了www.sencha.com ,但extjs这个域名会自动转到sencha上。
(2)帮助文档:
想要查看EXT的官方文档的话,可以去这里h
- mybatis3的mapper文件报Referenced file contains errors
xingguangsixian
mybatis
最近使用mybatis.3.1.0时无意中碰到一个问题:
The errors below were detected when validating the file "mybatis-3-mapper.dtd" via the file "account-mapper.xml". In most cases these errors can be d