2022.04.13【读书笔记】|10X单细胞转录组分析流程介绍
文章目录
- 摘要
- 课程目录
-
- 流程分析软件
- 基础分析流程
-
- 数据质控
-
- 测序统计
- 细胞过滤
- 表达定量
- 聚类分群
-
- 数据合并
- 批次效应
-
- MNN矫正
- CCA矫正
- harmony矫正
- 细胞过滤
- 亚群聚类
-
- 高变基因筛选与PCA分析
- 聚类
- 可视化
- 亚群鉴定(关键,耗时最久)
-
- 标记基因
- 亚群定义
- 特征分析
-
- 频率统计
- 亚群特征表达基因
- 个性数据挖掘
-
- 瞬时状态
- 细胞演化
- 细胞关联
- 小结
摘要
本次笔记是基迪奥单细胞课程第二章,根据课件以及上课老师的情况来看,比之前的美格和菲沙都要好一些,而且价格更优惠。从官网也可以看到基迪奥在培训这方面是作为重点业务。等上完剩余课程,我会进行一个简单的小结,来评估几个公司之间的课程质量。
之前公司生信团队已经对单细胞分析有部分研究讨论,本次课程可以对大家进行一个查缺补漏,内容也更加全面和体系化,因此还是比较推荐。
课程目录
流程分析软件
| 软件 | 期刊 | 运行环境 |
|---|---|---|
| CellRanger | - | - |
| Seurat | Nature Biotechnology | R |
| Scanpy | Genomic Biology | python |
| scVI | Nature Methods | python |
| Bioconductor | Nature Methods | R |
| …… |
基础分析流程
数据质控
测序统计
统计测序饱和度、捕获细胞数、平均数据量、平均检测基因数。
使用官方软件:Cell Ranger

细胞过滤
1)高RNA量细胞捕获
UMI总量最高的前N(预期细胞数)个细胞RNA总表达丰度的
99%分位数的1/10,作为有效细胞的最低阈值
2)低RNA量细胞捕获
确定空载的GEM的丰度基线,然后找出与空载的GEM存在显著差异
的GEM。这个步骤可能可以额外找到低RNA丰度的细胞。

表达定量
Read1 提 取 16bp GemCode Barcode 和12bp UMI,
Read2 提 取 cDNA插入片段
利用GemCode序列来区分细胞
利用UMI来区分不同原始转录本:利 用Read2 比对基因,
利用Read1 的UMI进行定量

聚类分群
数据合并
同时分析多个样本时,需要将各个样本合并为一套数据,便于
后期进行样本间的比较。
批次效应
批次效应(Batch effect)通常指的是实验指标检测中,来源关注的生物学处理效应之外的其他因素导致的样本结果的波动。
比如所检测的样本来源不同的实验环境、不同检测技术、试剂批次变化、不同实验员手法的差异,都会额外引入差异。
批次效应理论上在任何实验中都可能存在。高通量测序由于检测精度高,因此对批次效应更加敏感。

批次效应带来的基因表达变化,对任何定量研究都有影响。
• 10x单细胞研究的额外影响:导致本该聚类在一起的细胞因为批次效应被分为不同的簇,影响了细胞亚群鉴定的准确性,以及下游的所有分析。
实验解决方法:
- 有条件,就尽可能一次收集所有样本,一次做完(但实际情况往往不允许)
2)采用速冻法保存样本,等收集足够以后,用提核法一次完成文库制备(样本难收集,周期长的项目,可以考虑这种方法)。
生信解决方法:
MNN矫正
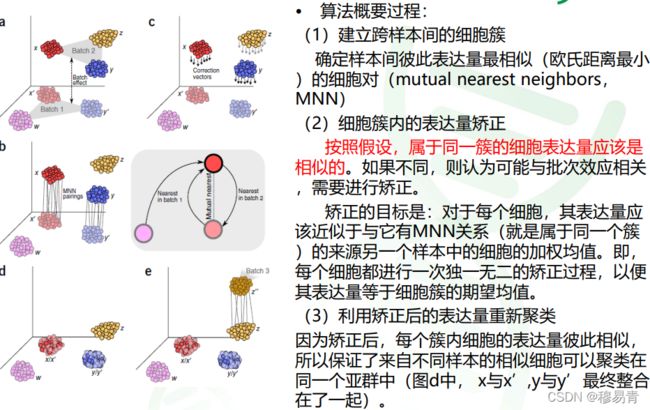
CCA矫正
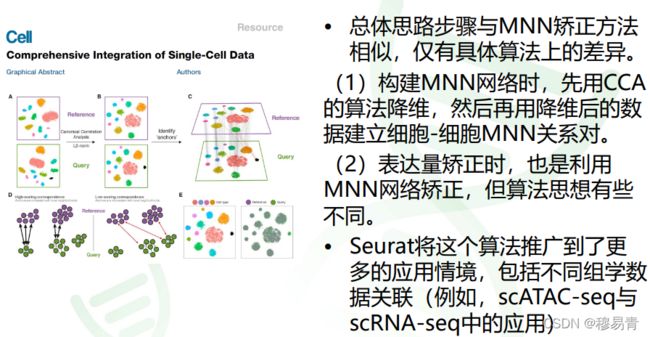
实操矫正代码
#####代码例子:
## We then identify anchors using the FindIntegrationAnchors function,
which takes a list of Seurat objects as input, and use these anchors to integrate
the two datasets together with IntegrateData.
#(1)# 建立MNN关系对
immune.anchors <- FindIntegrationAnchors(object.list =
ifnb.list, anchor.features = features)
# (2)对表达量进行矫正
immune.combined <- IntegrateData(anchorset = immune.anchors)
#(3)后续的聚类分群、差异分析等与正常流程类似
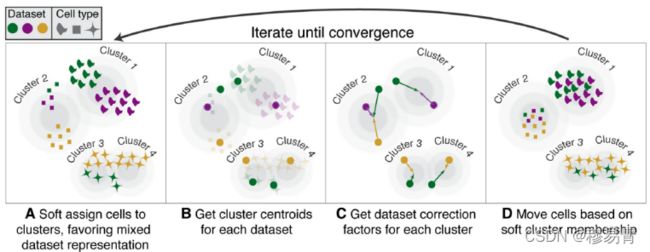
harmony矫正
首先,Harmony应用主成分分析,将转录组表达谱嵌入到低维空间中,然后应用迭代过程去除数据集特有的影响。
(A)Harmony概率性地将细胞分配给cluster,从而使每个cluster内数据
集的多样性最大化。
(B)Harmony计算每个cluster的所有数据集的全局中心,以及特定数据
集的中心。
(C)在每个cluster中,Harmony基于中心为每个数据集计算校正因子。
(D)最后,Harmony使用基于C的特定于细胞的因子校正每个细胞。由
于Harmony使用软聚类,因此可以通过多个因子的线性组合对其A中进行
的软聚类分配进行线性校正,来修正每个单细胞。
重复步骤A到D,直到收敛为止。聚类分配和数据集之间的依赖性随着每一
轮的减少而减小。
harmony算法优势
- 整合数据的同时对稀有细胞的敏感性依然很好;
- 省内存,运行速度快,适用于大样本;
- 适合于更复杂的单细胞分析实验设计,可以比较来自不同供体,组织和技术平台的细胞。
细胞过滤
细胞过滤是在开始正式分析前,进一步过滤得到可信的细胞用于
后续分析。
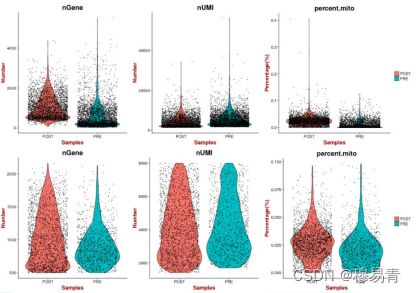
人为设定的一些经验标准,例如:
单细胞中鉴定到的gene数量
单细胞中UMI的总数。UMI总数过高则可能是由于实验过程中两个细胞进入了一个微滴,这类数据需要去除。
单细胞中UMI的线粒体基因表达量比例(小于10%,数值不固定)
亚群聚类
算法步骤:
(1)表达量均一化:A = log( 1 + ( UMIA ÷ UMITotal ) × 10000 )
其中,A:目标细胞中目标基因A的表达量;UMIA: 目标细胞中A基因的UMI数量;UMITotal:目标细胞中所有UMI数量的总和;log:以e为底数的自然对数。本质上就是消除不同细胞数据量的不同(因此,cell ranger的深度均一化是非必须的)。
(2)PCA分析,挑选主成分(降维),涉及:高可变基因筛选;
(3)聚类分群,涉及:分群标准的确定;
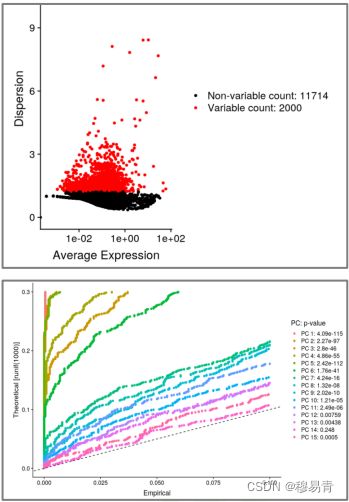
高变基因筛选与PCA分析
筛选方差较高的可变基因(例如top 2000)
PCA分析,并挑选显著(P<0.01)的主成分(PC),用于下一步的分析(降维)
本质:下一步用主成分进行亚群划分。因此主成分可以降低细胞间噪音信号的干扰。
聚类
Seurat软件使用基于图论的聚类算法对细胞进行聚类和分群。主要
包括以下步骤:
(1)构建细胞间的聚类关系:利用显著的主成分构建基于欧式距离的
KNN聚类关系图;
(2)优化细胞间聚类关系距离的权重值:利用Jaccard相似性优化细胞间
距离的权重值;
(3)聚类和分群:使用Louvain 算法进行细胞群聚类优化。
可视化
在群体单细胞领域,tSNE是更适合PCA的算法
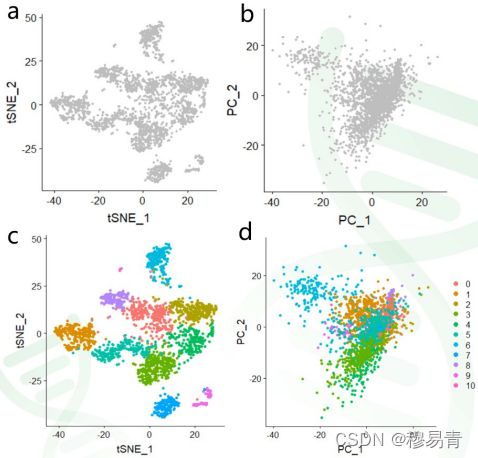
- 在这里t-SNE(t-Distributed Stochastic Neighbor Embedding) 只是用于将细胞排在一个二维空间里(散点图) 。然后将每个点根据亚群来源(上一步分析得到的),涂上不同的颜色。
- 为什么用tSNE?
答: tSNE是非线性的方法。简单理解就是:相似的细胞保持紧密聚类的同时,差异越大的细胞,在图中的距离越被夸大。这样不同亚群的细胞,群间的隔离更彻底,更清晰。还有类似的算法UMAP。 - 为什么图中相同颜色相同的细胞(属于一类)好像来源不同的簇?
答:因为分类定义来自上一页的步骤,而这里t-SNE聚类是另一种方法。不同方法,最后呈现出的细胞的相似性并不相同。
亚群鉴定(关键,耗时最久)
10X 单细胞转录组数据解读中,最耗费精力也是最关键的一部分是亚群的定义。
可以参考的鉴定步骤:
(1)从已有研究基础,样本特性等预估样本中潜在有哪些细胞亚群;
(2)搜寻对应细胞亚群的已知基因标记;
(3)基于已知基因标记进行亚群分类识别;
(4)对于依然无法定义的细胞亚群,也可以直接从亚群上调表达的基因去推测其潜在类型。或者,可以考虑,从其他其他物种/组织文献的相关报道中推测
标记基因
标记基因:又称marker基因,就是每个亚群特征或上调表达的基因(默认是和其他所有亚群比较)
一般来源文献或数据库
| 数据库 | 物种 | 细胞类型 | 数据信息 |
|---|---|---|---|
| Mouse cell Atlas | 小鼠 | 所有 | 基因 细胞亚群 |
| Cell Marker | 人、小鼠 | 所有 | 基因 细胞类型 文献出处 |
| Cancer SEA | 人 | 癌细胞 | 基因 癌症类型 功能解析 |
| PanglaoDB | 人、小鼠 | 所有 | 基因 细胞类型 统计学分析 |
| haemosphere | 人、小鼠 | 体液细胞 | 基因 细胞类型 表达量数 |
亚群定义
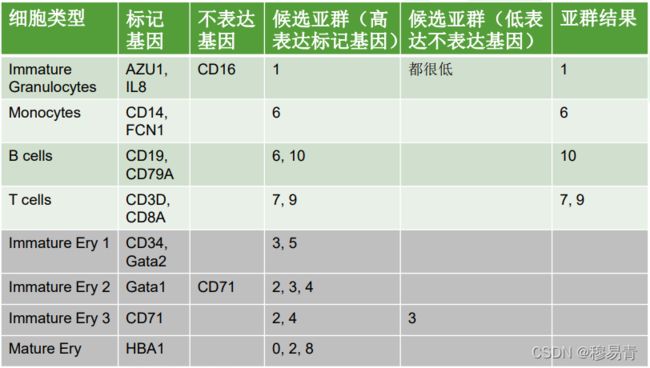
综合考虑各种逻辑,定义有争议的亚群(可以被定义到两类细胞的亚群)。
例如,本次范例红细胞相关的4个亚群,根据标记分群存在重叠。
(a) 满足IE2标记(表达的基因与不表达基因)要求的亚群只有C3,则先确定C3属于IE2;
(b) IE1只能选择C5;
© IE3(2,4)与ME(0,2,8)存在C2的重叠。无论从基因表达量还是聚类图中的距离,都很难判定,C2应该属于IE3还是ME,可能需要更多标记来判定。考虑到C2主要存在pre样本,暂时把他定义为成熟的细胞,所以分类为:
IE3:C4
ME:C0,C2,C8

特征分析
频率统计
细胞亚群频率(占总体百分比)的变化,也是常见的讨论内容。
这也是 10X genomics 设置生物学重复的主要意义之一。
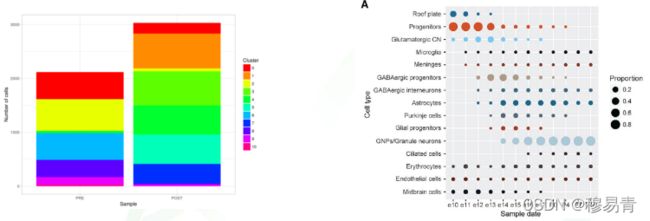
亚群特征表达基因
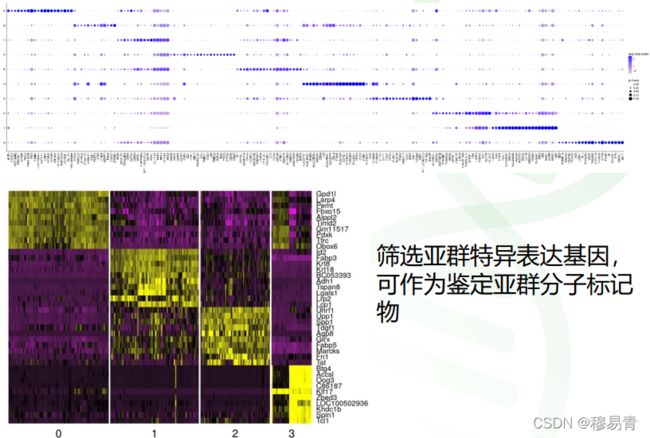
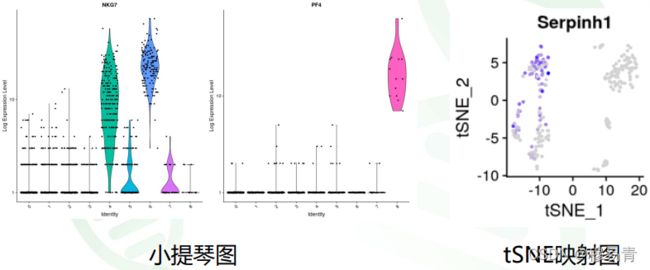
亚群特征基因的展示,经常使用小提琴图和映射图方式。

个性数据挖掘
瞬时状态
每个细胞都有独立的分子特征,了解细胞“此时此刻”所具有的特征是异质性研究的基础。
细胞功能(GSEA、GSVA)
拷贝数变异(CNV)
细胞演化
拟时分析
RNA速率分析
细胞周期分析
细胞关联
WGCNA分析
转录因子分析
细胞通讯分析
小结
关于标记基因
(1)特异标记基因,不等于特有表达。在其他类型的组织中也存在表达,只是相对丰度更低。甚至在其他细胞类型中,也存在较高表达。
(2)某个群体里的标记基因,并不意味着这个群体每个细胞都表达,而是平均表达量和表达所占的细胞比重较高。
有可能10X的结果与之前文献的报道不完全一致(文献解析中也会再次提及)。可能原因包括:由于样本状态(药物处理)、特定的组织时期、个体差异、检测技术不同等因素。
对单细胞有研究的小伙伴可以加微信bbplayer2021,进入交流群,一起交流研究。