R语言学习 - 热图简化
前面推出过热图绘制和热图美化,现在来一个函数绘制热图的简化方式。文后更有不用写代码的在线工具可用。
R语言 - 基础概念和矩阵操作
R语言 - 热图简化
R语言 - 热图绘制 (heatmap)
R语言 - 热图美化
热图绘制 - pheatmap
绘制热图除了使用ggplot2,还可以有其它的包或函数,比如pheatmap::pheatmap(pheatmap包中的pheatmap函数)、gplots::heatmap.2等。
相比于ggplot2作heatmap, pheatmap会更为简单一些,一个函数设置不同的参数,可以完成行列聚类、行列注释、Z-score计算、颜色自定义等。那我们来看看效果怎样。
data_ori <- "Grp_1;Grp_2;Grp_3;Grp_4;Grp_5
a;6.6;20.9;100.1;600.0;5.2
b;20.8;99.8;700.0;3.7;19.2
c;100.0;800.0;6.2;21.4;98.6
d;900;3.3;20.3;101.1;10000"
data <- read.table(text=data_ori, header=T, row.names=1, sep=";", quote="") Grp_1 Grp_2 Grp_3 Grp_4 Grp_5
a 6.6 20.9 100.1 600.0 5.2
b 20.8 99.8 700.0 3.7 19.2
c 100.0 800.0 6.2 21.4 98.6
d 900.0 3.3 20.3 101.1 10000.0pheatmap::pheatmap(data, filename="pheatmap_1.pdf")虽然有点丑,但一步就出来了。
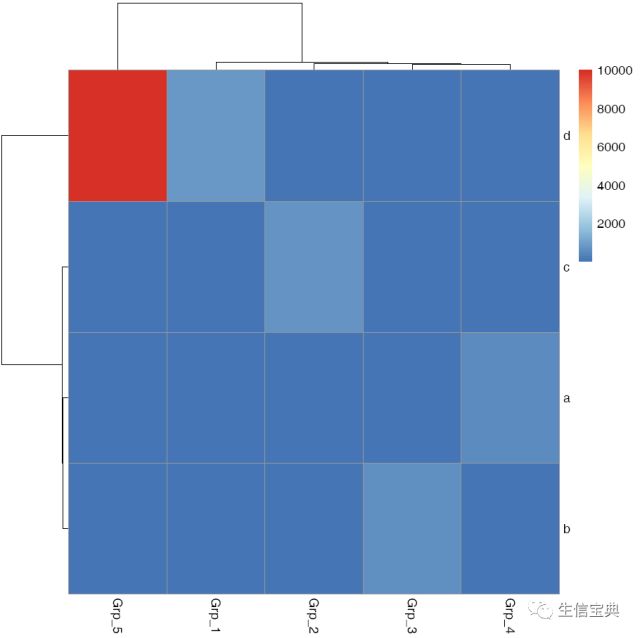 在
在heatmap美化篇提到的数据前期处理方式,都可以用于pheatmap的画图。此外Z-score计算在pheatmap中只要一个参数就可以实现。
pheatmap::pheatmap(data, scale="row", filename="pheatmap_1.pdf")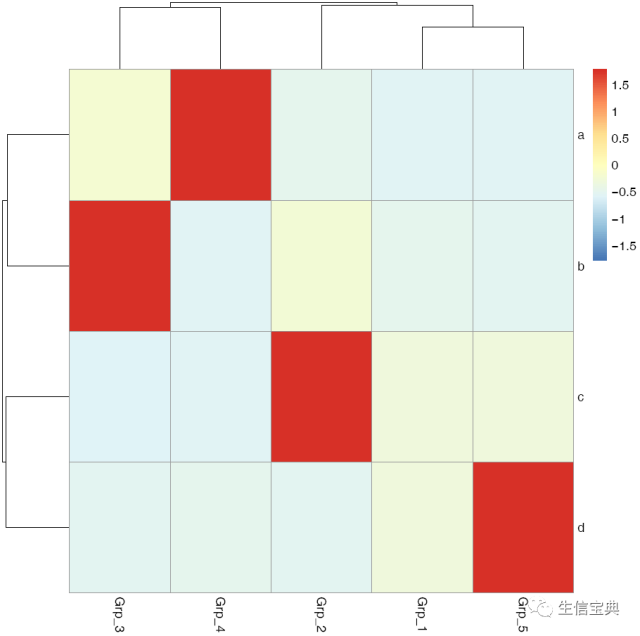
有时可能不需要行或列的聚类,原始展示就可以了。
pheatmap::pheatmap(data, scale="row", cluster_rows=FALSE, cluster_cols=FALSE, filename="pheatmap_1.pdf")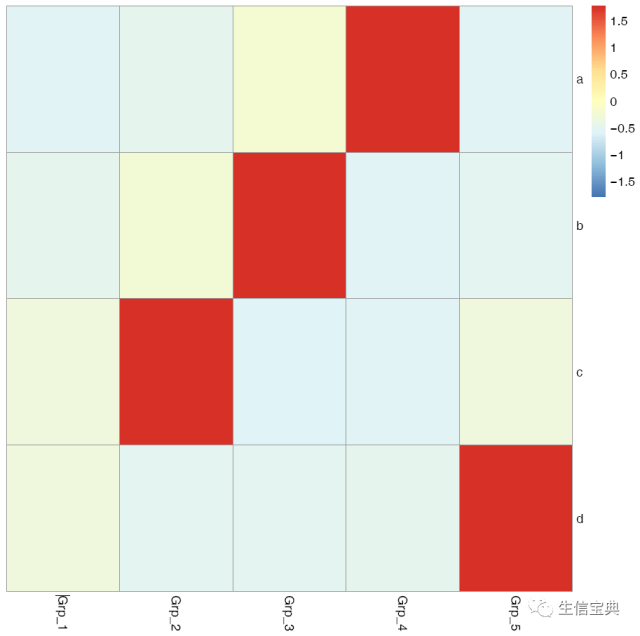
给矩阵 (data)中行和列不同的分组注释。假如有两个文件,第一个文件为行注释,其第一列与矩阵中的第一列内容相同 (顺序没有关系),其它列为第一列的不同的标记,如下面示例中(假设行为基因,列为样品)的2,3列对应基因的不同类型 (TF or enzyme)和不同分组。第二个文件为列注释,其第一列与矩阵中第一行内容相同,其它列则为样品的注释。
row_anno = data.frame(type=c("TF","Enzyme","Enzyme","TF"), class=c("clu1","clu1","clu2","clu2"), row.names=rownames(data))
row_anno type class
a TF clu1
b Enzyme clu1
c Enzyme clu2
d TF clu2col_anno = data.frame(grp=c("A","A","A","B","B"), size=1:5, row.names=colnames(data))
col_anno grp size
Grp_1 A 1
Grp_2 A 2
Grp_3 A 3
Grp_4 B 4
Grp_5 B 5pheatmap::pheatmap(data, scale="row",
cluster_rows=FALSE,
annotation_col=col_anno,
annotation_row=row_anno,
filename="pheatmap_1.pdf")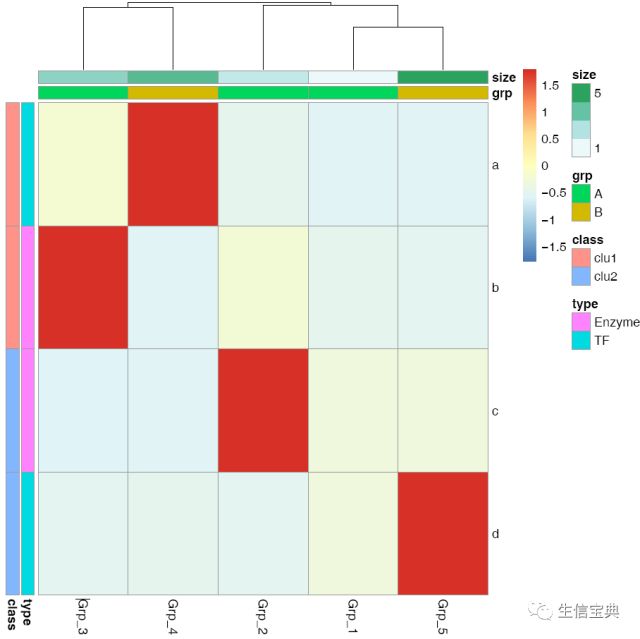
自定义下颜色吧。
# values larger than 1 will give more color for high end.
# Values between 0-1 will give more color for low end.
pheatmap::pheatmap(data, scale="row",
cluster_rows=FALSE,
annotation_col=col_anno,
annotation_row=row_anno,
color=colorRampPalette(c('green','yellow','red'), bias=1)(50),
filename="pheatmap_1.pdf") 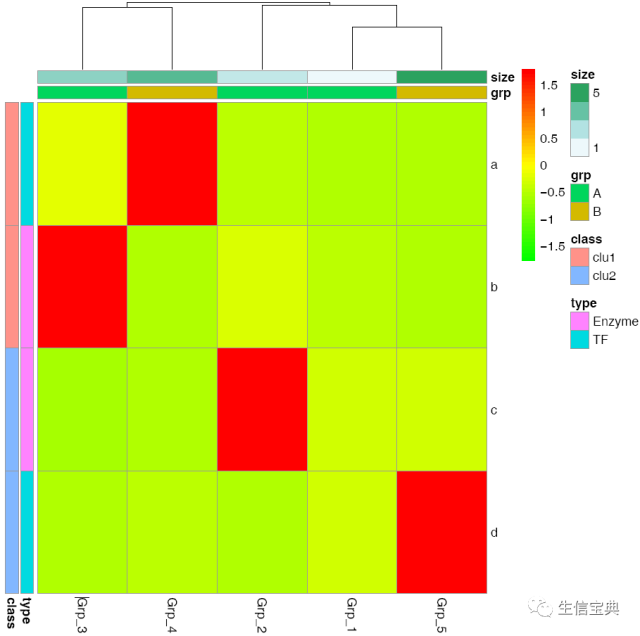
heatmap.2的使用就不介绍了,跟pheatmap有些类似,而且也有不少教程。
不改脚本的热图绘制
绘图时通常会碰到两个头疼的问题:
需要画很多的图,唯一的不同就是输出文件,其它都不需要修改。如果用R脚本,需要反复替换文件名,繁琐又容易出错。
每次绘图都需要不断的调整参数,时间久了不用,就忘记参数放哪了;或者调整次数过多,有了很多版本,最后不知道用哪个了。
为了简化绘图、维持脚本的一致,我用bash对R做了一个封装,然后就可以通过修改命令好参数绘制不同的图了。
先看一看怎么使用
首先把测试数据存储到文件中方便调用。数据矩阵存储在heatmap_data.xls文件中;行注释存储在heatmap_row_anno.xls文件中;列注释存储在heatmap_col_anno.xls文件中。
# tab键分割,每列不加引号
write.table(data, file="heatmap_data.xls", sep="\t", row.names=T, col.names=T,quote=F)
# 如果看着第一行少了ID列不爽,可以填补下
system("sed -i '1 s/^/ID\t/' heatmap_data.xls")
write.table(row_anno, file="heatmap_row_anno.xls", sep="\t", row.names=T, col.names=T,quote=F)
write.table(col_anno, file="heatmap_col_anno.xls", sep="\t", row.names=T, col.names=T,quote=F)然后用程序sp_pheatmap.sh绘图。
# -f: 指定输入的矩阵文件
# -d:指定是否计算Z-score, (否), (按行算),
一个回车就得到了下面的图
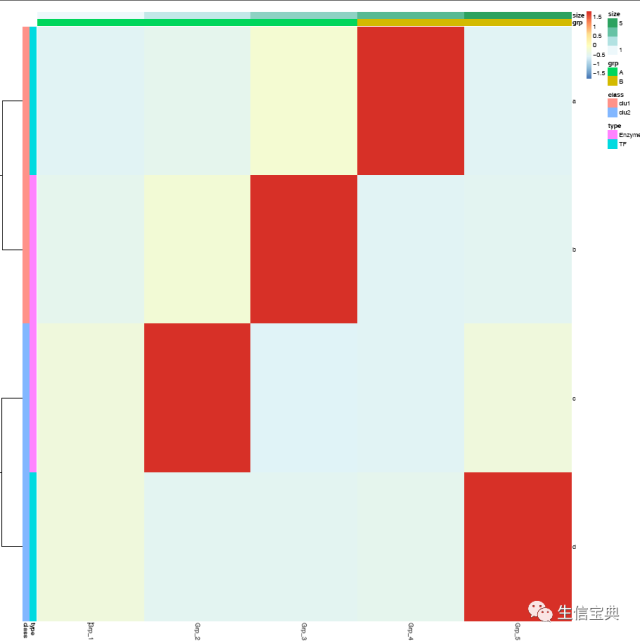
字有点小,是因为图太大了,把图的宽和高缩小下试试。
# -f: 指定输入的矩阵文件
# -d:指定是否计算Z-score, (否), (按行算),
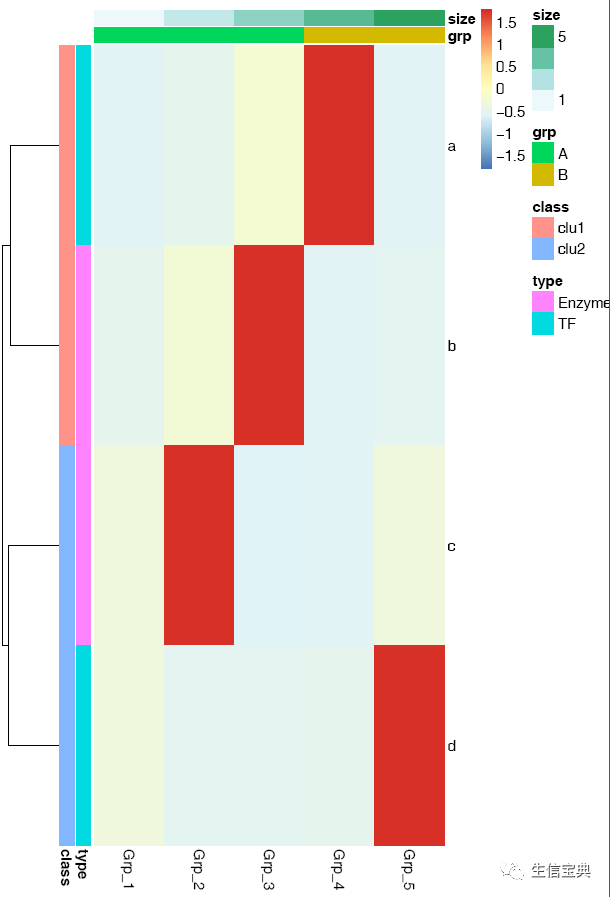
横轴的标记水平放置
# -A: 0, X轴标签选择0度
# -C: 自定义颜色,注意引号的使用,最外层引号与内层引号不同,引号之间无交叉
# -T: 指定给定的颜色的类型;如果给的是vector (如下面的例子), 则-T需要指定为vector; 否则结果会很怪异,只有俩颜色。
# -t: 指定图形的题目,注意引号的使用;参数中包含空格或特殊字符等都要用引号引起来作为一个整体。
ct@ehbio:~/$ sp_pheatmap.sh -f heatmap_data.xls -d row -P heatmap_row_anno.xls -Q heatmap_col_anno.xls -u 8 -v 12 -A 0 -C 'c("white", "blue")' -T vector -t "Heatmap of gene expression profile"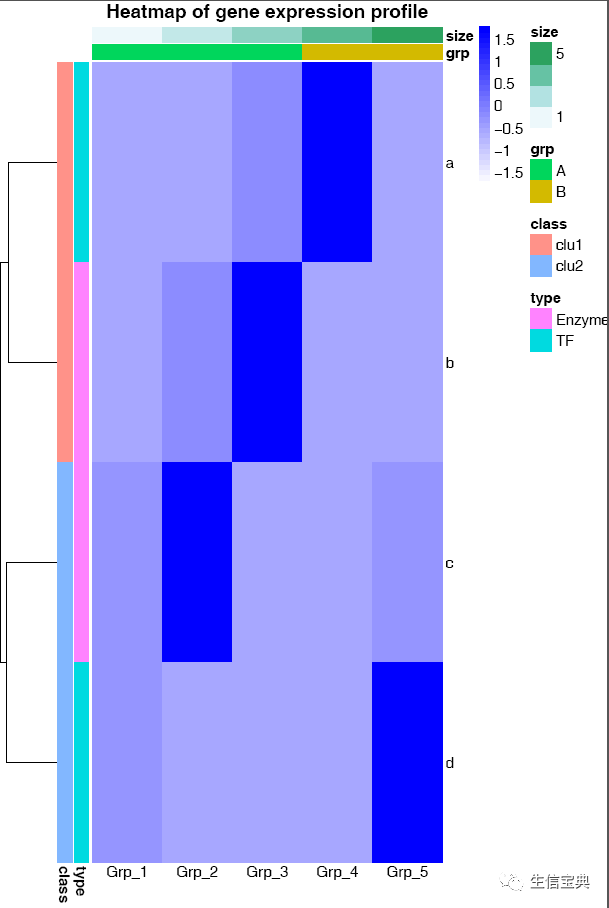
sp_pheatmap.sh的参数还有一些,可以完成前面讲述过的所有热图的绘制,具体如下:
***CREATED BY Chen Tong ([email protected])***
----Matrix file--------------
Name T0_1 T0_2 T0_3 T4_1 T4_2
TR19267|c0_g1|CYP703A2 1.431 0.77 1.309 1.247 0.485
TR19612|c1_g3|CYP707A1 0.72 0.161 0.301 2.457 2.794
TR60337|c4_g9|CYP707A1 0.056 0.09 0.038 7.643 15.379
TR19612|c0_g1|CYP707A3 2.011 0.689 1.29 0 0
TR35761|c0_g1|CYP707A4 1.946 1.575 1.892 1.019 0.999
TR58054|c0_g2|CYP707A4 12.338 10.016 9.387 0.782 0.563
TR14082|c7_g4|CYP707A4 10.505 8.709 7.212 4.395 6.103
TR60509|c0_g1|CYP707A7 3.527 3.348 2.128 3.257 2.338
TR26914|c0_g1|CYP710A1 1.899 1.54 0.998 0.255 0.427
----Matrix file--------------
----Row annorarion file --------------
------1. At least two columns--------------
------2. The first column should be the same as the first column in
matrix (order does not matter)--------------
Name Clan Family
TR19267|c0_g1|CYP703A2 CYP71 CYP703
TR19612|c1_g3|CYP707A1 CYP85 CYP707
TR60337|c4_g9|CYP707A1 CYP85 CYP707
TR19612|c0_g1|CYP707A3 CYP85 CYP707
TR35761|c0_g1|CYP707A4 CYP85 CYP707
TR58054|c0_g2|CYP707A4 CYP85 CYP707
TR14082|c7_g4|CYP707A4 CYP85 CYP707
TR60509|c0_g1|CYP707A7 CYP85 CYP707
TR26914|c0_g1|CYP710A1 CYP710 CYP710
----Row annorarion file --------------
----Column annorarion file --------------
------1. At least two columns--------------
------2. The first column should be the same as the first row in
---------matrix (order does not matter)--------------
Name Sample
T0_1 T0
T0_2 T0
T0_3 T0
T4_1 T4
T4_2 T4
----Column annorarion file --------------
Usage:
sp_pheatmap.sh options
Function:
This script is used to do heatmap using package pheatmap.
The parameters for logical variable are either TRUE or FALSE.
OPTIONS:
-f Data file (with header line, the first column is the
rowname, tab seperated. Colnames must be unique unless you
know what you are doing.)[NECESSARY]
-t Title of picture[Default empty title]
["Heatmap of gene expression profile"]
-a Display xtics. [Default TRUE]
-A Rotation angle for x-axis value (anti clockwise)
[Default 90]
-b Display ytics. [Default TRUE]
-H Hieratical cluster for columns.
Default FALSE, accept TRUE
-R Hieratical cluster for rows.
Default TRUE, accept FALSE
-c Clustering method, Default "complete".
Accept "ward.D", "ward.D2","single", "average" (=UPGMA),
"mcquitty" (=WPGMA), "median" (=WPGMC) or "centroid" (=UPGMC)
-C Color vector.
Default pheatmap_default.
Aceept a vector containing multiple colors such as
<'c("white", "blue")'> will be transferred
to
or an R function
generating a list of colors.
-T Color type, a vetcor which will be transferred as described in <-C> [vector] or
a raw vector [direct vector] or a function [function (default)].
-B A positive number. Default 1. Values larger than 1 will give more color
for high end. Values between 0-1 will give more color for low end.
-D Clustering distance method for rows.
Default 'correlation', accept 'euclidean',
"manhattan", "maximum", "canberra", "binary", "minkowski".
-I Clustering distance method for cols.
Default 'correlation', accept 'euclidean',
"manhattan", "maximum", "canberra", "binary", "minkowski".
-L First get log-value, then do other analysis.
Accept an R function log2 or log10.
[Default FALSE]
-d Scale the data or not for clustering and visualization.
[Default 'none' means no scale, accept 'row', 'column' to
scale by row or column.]
-m The maximum value you want to keep, any number larger willl
be taken as this given maximum value.
[Default Inf, Optional]
-s The smallest value you want to keep, any number smaller will
be taken as this given minimum value.
[Default -Inf, Optional]
-k Aggregate the rows using kmeans clustering.
This is advisable if number of rows is so big that R cannot
handle their hierarchical clustering anymore, roughly more than 1000.
Instead of showing all the rows separately one can cluster the
rows in advance and show only the cluster centers. The number
of clusters can be tuned here.
[Default 'NA' which means no
cluster, other positive interger is accepted for executing
kmeans cluster, also the parameter represents the number of
expected clusters.]
-P A file to specify row-annotation with format described above.
[Default NA]
-Q A file to specify col-annotation with format described above.
[Default NA]
-u The width of output picture.[Default 20]
-v The height of output picture.[Default 20]
-E The type of output figures.[Default pdf, accept
eps/ps, tex (pictex), png, jpeg, tiff, bmp, svg and wmf)]
-r The resolution of output picture.[Default 300 ppi]
-F Font size [Default 14]
-p Preprocess data matrix to avoid 'STDERR 0 in cor(t(mat))'.
Lowercase .
[Default TRUE]
-e Execute script (Default) or just output the script.
[Default TRUE]
-i Install the required packages. Normmaly should be TRUE if this is
your first time run s-plot.[Default FALSE]
sp_pheatmap.sh是我写作的绘图工具s-plot的一个功能,s-plot可以绘制的图的类型还有一些,列举如下;在后面的教程中,会一一提起。
Usage:
s-plot options
Function:
This software is designed to simply the process of plotting and help
researchers focus more on data rather than technology.
Currently, the following types of plot are supported.
#### Bars
s-plot barPlot
s-plot horizontalBar
s-plot multiBar
s-plot colorBar
#### Lines
s-plot lines
#### Dots
s-plot pca
s-plot scatterplot
s-plot scatterplot3d
s-plot scatterplot2
s-plot scatterplotColor
s-plot scatterplotContour
s-plot scatterplotLotsData
s-plot scatterplotMatrix
s-plot scatterplotDoubleVariable
s-plot contourPlot
s-plot density2d
#### Distribution
s-plot areaplot
s-plot boxplot
s-plot densityPlot
s-plot densityHistPlot
s-plot histogram
#### Cluster
s-plot hcluster_gg (latest)
s-plot hcluster
s-plot hclust (depleted)
#### Heatmap
s-plot heatmapS
s-plot heatmapM
s-plot heatmap.2
s-plot pheatmap
s-plot pretteyHeatmap # obseleted
s-plot prettyHeatmap
#### Others
s-plot volcano
s-plot vennDiagram
s-plot upsetView为了推广,也为了激起大家的热情,如果想要sp_pheatmap.sh脚本的,还需要劳烦大家动动手,转发此文章到朋友圈,并后台回复 s-plot 索取。
易生信系列培训课程,扫码获取免费资料

更多阅读
画图三字经 生信视频 生信系列教程
心得体会 TCGA数据库 Linux Python
高通量分析 免费在线画图 测序历史 超级增强子
生信学习视频 PPT EXCEL 文章写作 ggplot2
海哥组学 可视化套路 基因组浏览器
色彩搭配 图形排版 互作网络
自学生信
后台回复“生信宝典福利第一波”获取教程合集
高颜值免费在线绘图
R统计和作图
Graphpad,经典绘图工具初学初探
在R中赞扬下努力工作的你,奖励一份CheatShet
别人的电子书,你的电子书,都在bookdown
R语言 - 入门环境Rstudio
R语言 - 热图绘制 (heatmap)
R语言 - 基础概念和矩阵操作
R语言 - 热图简化
R语言 - 热图美化
R语言 - 线图绘制
R语言 - 线图一步法
R语言 - 箱线图(小提琴图、抖动图、区域散点图)
R语言 - 箱线图一步法
R语言 - 火山图
R语言 - 富集分析泡泡图
R语言 - 散点图绘制
R语言 - 韦恩图
R语言 - 柱状图
R语言 - 图形设置中英字体
R语言 - 非参数法生存分析
R语言 - 绘制seq logo图
WGCNA分析,简单全面的最新教程
psych +igraph:共表达网络构建
一文学会网络分析——Co-occurrence网络图在R中的实现
一文看懂PCA主成分分析
富集分析DotPlot,可以服
基因共表达聚类分析和可视化
R中1010个热图绘制方法
还在用PCA降维?快学学大牛最爱的t-SNE算法吧, 附Python/R代码
一个函数抓取代谢组学权威数据库HMDB的所有表格数据
文章用图的修改和排版
network3D: 交互式桑基图
network3D 交互式网络生成
Seq logo 在线绘制工具——Weblogo
生物AI插图素材获取和拼装指导
ggplot2高效实用指南 (可视化脚本、工具、套路、配色)
图像处理R包magick学习笔记
SOM基因表达聚类分析初探
利用gganimate可视化全球范围R-Ladies(R社区性别多样性组织)发展情况
一分钟绘制磷脂双分子层:AI零基础入门和基本图形绘制
AI科研绘图(二):模式图的基本画法
你知道R中的赋值符号箭头(<-)和等号(=)的区别吗?
R语言可视化学习笔记之ggridges包
利用ComplexHeatmap绘制热图(一)
ggplot2学习笔记之图形排列
R包reshape2,轻松实现长、宽数据表格转换
用R在地图上绘制网络图的三种方法
PCA主成分分析实战和可视化 附R代码和测试数据
iTOL快速绘制颜值最高的进化树!
12个ggplot2扩展包帮你实现更强大的可视化
编程模板-R语言脚本写作:最简单的统计与绘图,包安装、命令行参数解析、文件读取、表格和矢量图输出
R语言统计入门课程推荐——生物科学中的数据分析Data Analysis for the Life Sciences
数据可视化基本套路总结
你知道R中的赋值符号箭头
<-和等号=的区别吗?使用dplyr进行数据操作30例
交集intersect、并集union、找不同setdiff
R包reshape2,轻松实现长、宽数据表格转换
1数据类型(向量、数组、矩阵、 列表和数据框)
2读写数据所需的主要函数、与外部环境交互
3数据筛选——提取对象的子集
4向量、矩阵的数学运算
5控制结构
6函数及作用域
7认识循环函数lapply和sapply
8分解数据框split和查看对象str
9模拟—随机数、抽样、线性模型
1初识ggplot2绘制几何对象
2图层的使用—基础、加标签、注释
3工具箱—误差线、加权数、展示数据分布
4语法基础
5通过图层构建图像
6标度、轴和图例
7定位-分面和坐标系
8主题设置、存储导出
9绘图需要的数据整理技术
创建属于自己的调色板
28个实用绘图包,总有几个适合你
热图绘制
R做线性回归
绘图相关系数矩阵corrplot
相关矩阵可视化ggcorrplot
绘制交互式图形recharts
交互式可视化CanvasXpress
聚类分析factoextra
LDA分析、作图及添加置信-ggord
解决散点图样品标签重叠ggrepel
添加P值或显著性标记ggpubr
Alpha多样性稀释曲线rarefraction curve
堆叠柱状图各成分连线画法:突出组间变化
冲击图展示组间时间序列变化ggalluvial
桑基图riverplot
微生物环境因子分析ggvegan
五彩进化树与热图更配ggtree
多元回归树分析mvpart
随机森林randomForest 分类Classification 回归Regression
加权基因共表达网络分析WGCNA
circlize包绘制circos-plot
R语言搭建炫酷的线上博客系统
28个实用绘图包,总有几个适合你
Cytoscape教程1
Cytoscape之操作界面介绍
新出炉的Cytoscape视频教程
Cytoscape制作带bar图和pie图节点的网络图
Cytoscape: MCODE增强包的网络模块化分析
听说分享到朋友圈的朋友会在公众号周年庆时中奖 (大家还记得去年的大放送吧,不记得查查历史 )
)

学习宏基因组关照此公众号。