增强火山图,要不要试一下?
包作者:Kevin Blighe
撰文:协和医学院 苑晓梅
编辑:生信宝典
前言
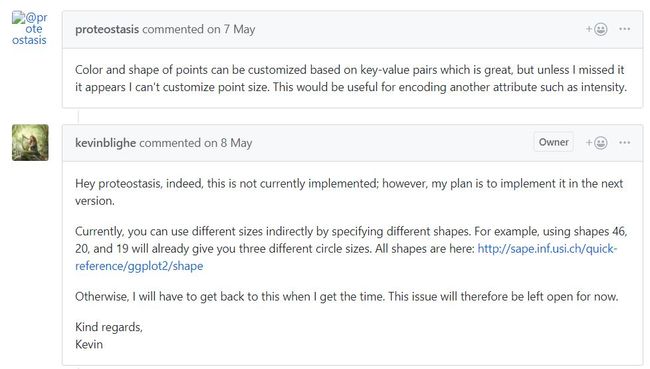
最近道听途说EnhancedVolcano绘制火山图的方便性,所以本人就根据其说明文档进行操作。但在操作过程中发现,其shape功能并没有在help文档中找到,经过搜索在github上看到了以下的答复 。。。(说明整个文档功能并没有完全开发,需进行选择)
1 Introduction
火山图是可视化差异表达分析结果的有效方法。这次更新的EnhancedVolcano目的就是两个(1)使转录本基因名称的显示更加的合理化,避免出现相互重叠的现象;(2)允许用户通过颜色,形状和阴影参数配置在同一绘图空间中识别多达3种不同类型的属性。
2 Installation
2.1 1. 下载安装包
# if (!requireNamespace('BiocManager', quietly = TRUE))
# install.packages('BiocManager')
# BiocManager::install('EnhancedVolcano')
if (!requireNamespace('devtools', quietly = TRUE))
install.packages('devtools')
devtools::install_github('kevinblighe/EnhancedVolcano')2.2 2. 加载R包
library(EnhancedVolcano)3 开始
作者使用该流程: RNA-seq workflow: gene-level exploratory analysis and differential expression。具体来说,我们将加载airway数据,其中不同的气道平滑肌细胞用地塞米松治疗。
library(airway)
library(magrittr)
data('airway')
# %<>%复合赋值操作符, 功能与 %>% 基本是一样的,但多了一项额外的操作,就是把结果写到左侧对象。
# 对dex列进行relevel,再把revel后的结果赋值到airway$dex。
airway$dex %<>% relevel('untrt')使用DESeq2进行差异表达,以创建两组结果(DESeq2差异基因分析和批次效应移除):
library('DESeq2')
dds <- DESeqDataSet(airway, design = ~ cell + dex)
dds <- DESeq(dds, betaPrior=FALSE)
# compare trt & untrt
res1 <- results(dds,
contrast = c('dex','trt','untrt'))
# shrink log2 fold change
res1 <- lfcShrink(dds,
contrast = c('dex','trt','untrt'), res=res1)
# compare different cells
res2 <- results(dds,
contrast = c('cell', 'N061011', 'N61311'))
res2 <- lfcShrink(dds,
contrast = c('cell', 'N061011', 'N61311'), res=res2)查看下数据结构
head res1log2 fold change (MAP): dex trt vs untrt
Wald test p-value: dex trt vs untrt
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE
ENSG00000000003 708.602169691234 -0.374152710396614 0.0988428916720785
ENSG00000000005 0 NA NA
ENSG00000000419 520.297900552084 0.202062036081026 0.109739490807055
ENSG00000000457 237.163036796015 0.0361672062398394 0.138337785736641
ENSG00000000460 57.9326331250967 -0.0844566831590659 0.249890471495246
ENSG00000000938 0.318098378392895 -0.0841390331826692 0.151334283397515
stat pvalue padj
ENSG00000000003 -3.7877506903658 0.000152017272634539 0.00128363812227422
ENSG00000000005 NA NA NA
ENSG00000000419 1.84294384315416 0.0653372100766985 0.19654584069126
ENSG00000000457 0.264356843264039 0.791504963002101 0.911458000845921
ENSG00000000460 -0.307052600205469 0.758803335537917 0.895034449952733
ENSG00000000938 -0.39379516719652 0.693732272741941 NA 3.1 绘制最基本的火山图
对于最基本的火山图,只需要一个数据框或测试结果矩阵,包含转录本名称,log2FC以及adjusted或unajusted的P值。 log2FC的默认cut-off值是 > | 2 |; P值的默认cut-off值为10e-6。
EnhancedVolcano(res1,
# 基因名字
lab = rownames(res1),
x = 'log2FoldChange',
y = 'pvalue',
xlim = c(-5, 8))图例:NS-非显著基因;Log2 FC倍数大于阈值的基因;P 统计显著的基因;P & Log2 FC 差异基因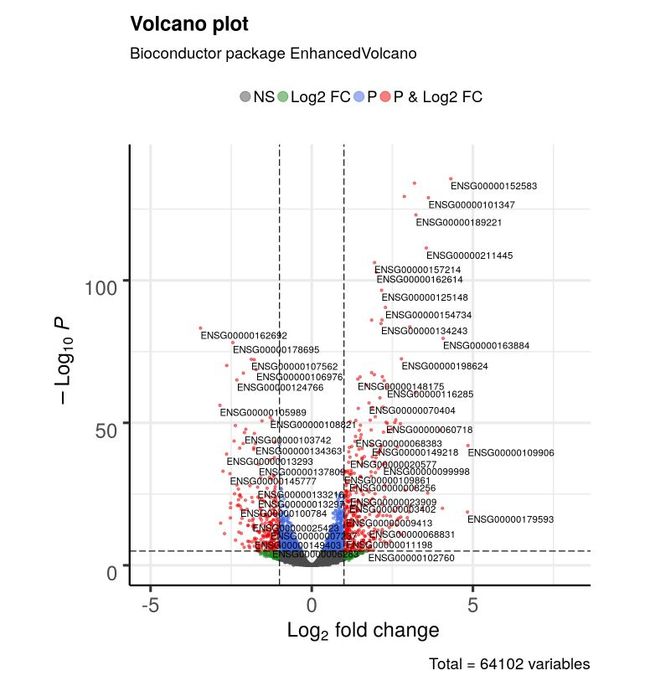
4 高级功能
默认情况下,EnhancedVolcano将仅尝试标记设置的阈值筛选出的差异基因,即p Cutoff和FC cutoff。 此外,它只会标记可以合理地适合绘图空间的基因。 用户可以选择性地提供他/她希望在图中标记的转录本名称的矢量(as selectLab)。
在这个例子中,还修改了点和标签大小,帮助改善清晰度,保障更多的转录本进入差异分析中。
EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
xlim = c(-8, 8),
title = 'N061011 versus N61311',
pCutoff = 10e-16,
FCcutoff = 1.5,
transcriptPointSize = 1.5,
transcriptLabSize = 3.0)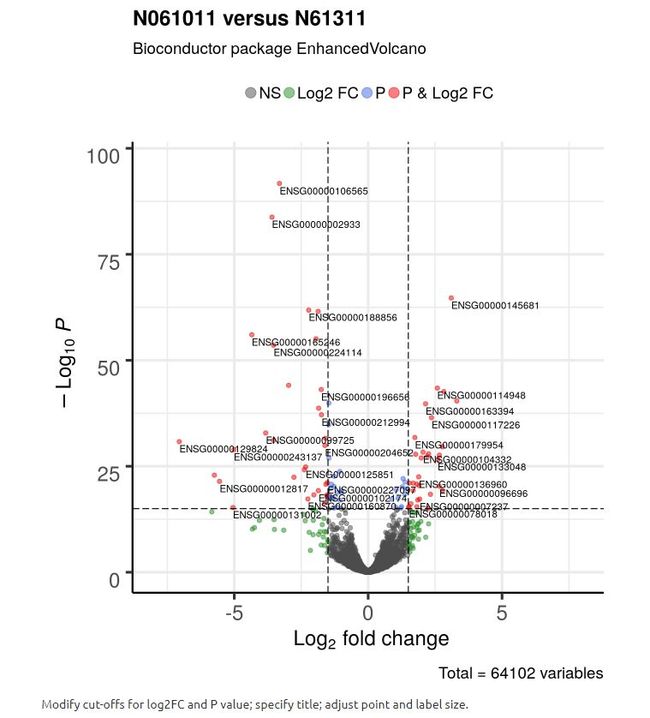
4.2 调整点的颜色和透明度
默认配色方案可能不是每个人都喜欢。 在这里,只有通过log2FC和P值筛选的差异转录本都是红色的,其他一切都是黑色的。 还调整’alpha’的值,它控制绘制点的透明度:1 = 100%不透明; 0 = 100%透明。
EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
xlim = c(-8, 8),
title = 'N061011 versus N61311',
pCutoff = 10e-16,
FCcutoff = 1.5,
transcriptPointSize = 1.5,
transcriptLabSize = 3.0,
# Colour shading for plotted points, corresponding to < abs(FCcutoff) && > pCutoff,
# > abs(FCcutoff), < pCutoff, > abs(FCcutoff) && < pCutoff.
# 无显著,倍数大(左下、右下),P小 (中上), 显著差异
# > DEFAULT = c("grey30", "forestgreen", "royalblue", "red2").
col=c('black', 'black', 'black', 'red3'),
colAlpha = 1)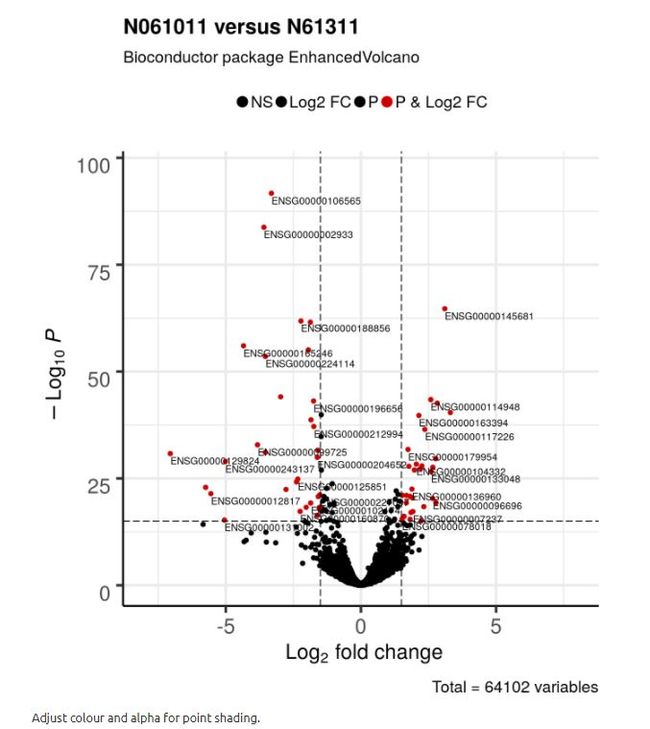
4.3 调整绘制点的形状
它可以帮助将不同的点绘制成不同的形状。 默认形状是圆形。 用户可以通过shape参数指定形状,该参数接受单个或四个可能的值:如果有四个值,则这些值将映射到也由颜色指定的标准名称; 如果是单个值,则所有点都用此值绘制。
For more information on shape encoding search online at ggplot2 Quick Reference: shape
EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
xlim = c(-8, 8),
title = 'N061011 versus N61311',
pCutoff = 10e-16,
FCcutoff = 1.5,
transcriptPointSize = 3.0,
transcriptLabSize = 3.0,
shape = 8,
colAlpha = 1)
# 注意Bioconductor版本该处shape功能并不能用,需要安装github的开发版![]()
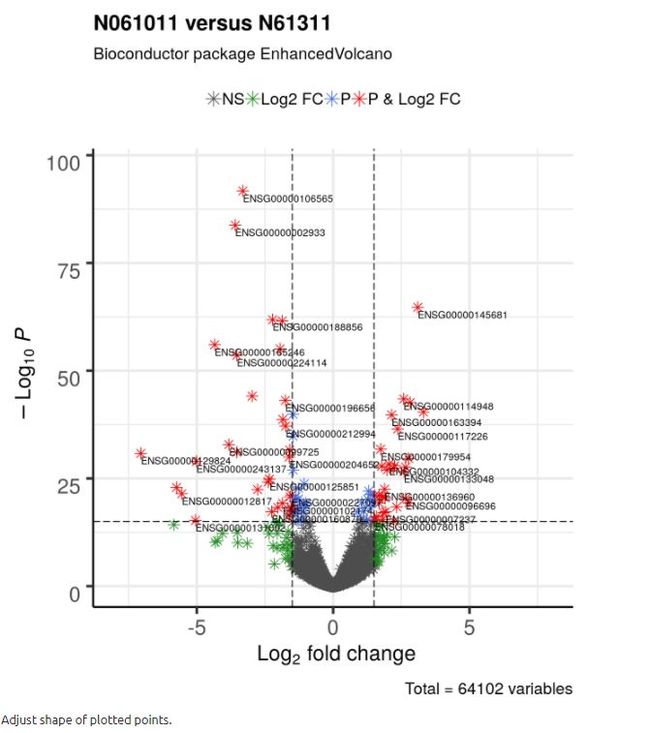
调整画图点的形状
EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
xlim = c(-8, 8),
title = 'N061011 versus N61311',
pCutoff = 10e-16,
FCcutoff = 1.5,
transcriptPointSize = 2.0,
transcriptLabSize = 3.0,
# 同上面col
# 无显著,倍数大(左下、右下),P小 (中上), 显著差异
shape = c(1, 4, 23, 25),
colAlpha = 1)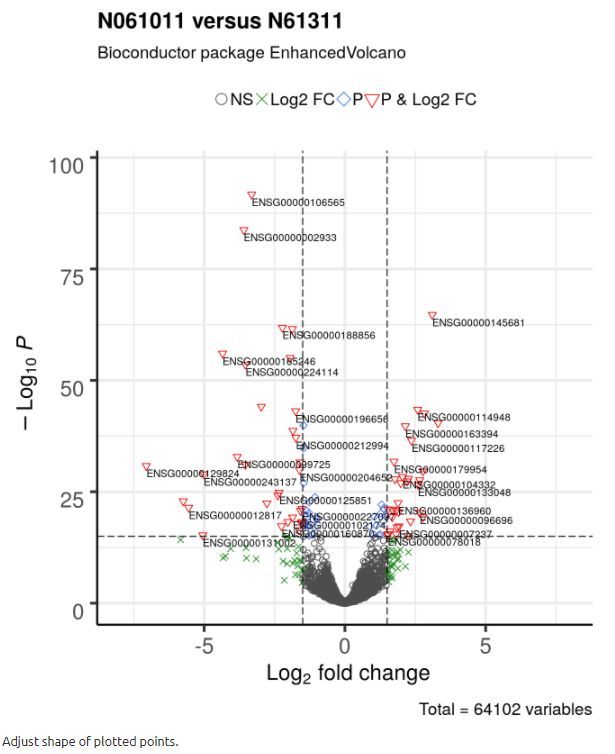
4.4 调整cut-off线并添加额外的阈值线
cut-off线可以通过以下参数进行调整。 “cutoffLineType”以下参数进行修改:“blank”, “solid”, “dashed”, “dotted”, “dotdash”, “longdash”, “twodash”;cutoff线的颜色和粗细可以通过 ‘cutoffLineCol’ 和 ‘cutoffLineWidth’进行修改,如果不需要该cut-off线,可以设置“cutoffLineType=“blank” or cutoffLineWidth=0.”
也可以通过参数‘hline’ and ‘vline’ 显示其他的cut-off线;
EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
xlim = c(-6, 6),
title = 'N061011 versus N61311',
pCutoff = 10e-12,
FCcutoff = 1.5,
transcriptPointSize = 1.5,
transcriptLabSize = 3.0,
colAlpha = 1,
# 取消cutoff线
cutoffLineType = 'blank',
cutoffLineCol = 'black',
cutoffLineWidth = 0.8,
hline = c(10e-12, 10e-36, 10e-60, 10e-84),
hlineCol = c('grey0', 'grey25','grey50','grey75'),
hlineType = 'longdash',
hlineWidth = 0.8,
gridlines.major = FALSE,
gridlines.minor = FALSE)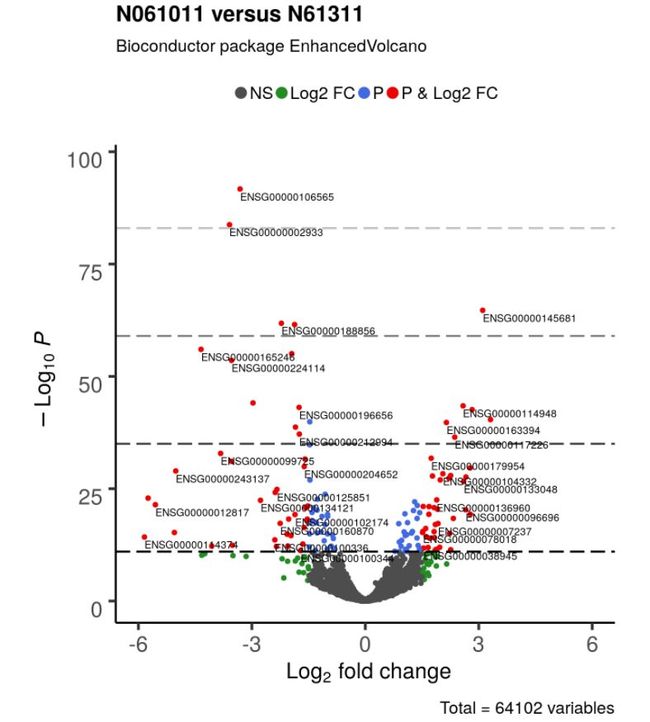
4.5 调整图例位置,大小和文本
EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
xlim = c(-6, 6),
pCutoff = 10e-12,
FCcutoff = 1.5,
cutoffLineType = 'twodash',
cutoffLineWidth = 0.8,
transcriptPointSize = 3.0,
transcriptLabSize = 4.0,
colAlpha = 1,
legend=c('NS','Log (base 2) fold-change','P value', 'P value & Log (base 2) fold-change'),
legendPosition = 'right',
legendLabSize = 16,
legendIconSize = 5.0)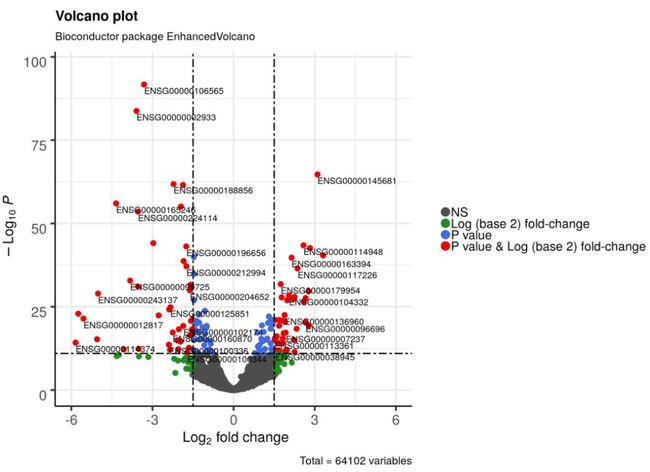
4.6 绘制调整后的p值
作者通过 bquote 函数修改轴标题
EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'padj',
xlim=c(-6,6),
xlab = bquote(~Log[2]~ 'fold change'),
ylab = bquote(~-Log[10]~adjusted~italic(P)),
pCutoff = 0.0001,
FCcutoff = 1.0,
transcriptLabSize = 4.0,
colAlpha = 1,
legend=c('NS','Log2 FC','Adjusted p-value','Adjusted p-value & Log2 FC'),
legendPosition = 'bottom',
legendLabSize = 10,
legendIconSize = 3.0)4.7 通过添加连接线来添加更多标签
为了标记更多点,可以通过短线连接点和标签, 这些连接线的宽度和颜色也可以分别用widthConnectors和colConnectors进行修改;
EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
xlim = c(-6,6),
xlab = bquote(~Log[2]~ 'fold change'),
pCutoff = 10e-14,
FCcutoff = 2.0,
transcriptPointSize = 3.0,
transcriptLabSize = 4.0,
colAlpha = 1,
legend=c('NS','Log (base 2) fold-change','P value', 'P value & Log (base 2) fold-change'),
legendPosition = 'right',
legendLabSize = 12,
legendIconSize = 4.0,
drawConnectors = TRUE,
widthConnectors = 0.2,
colConnectors = 'grey30')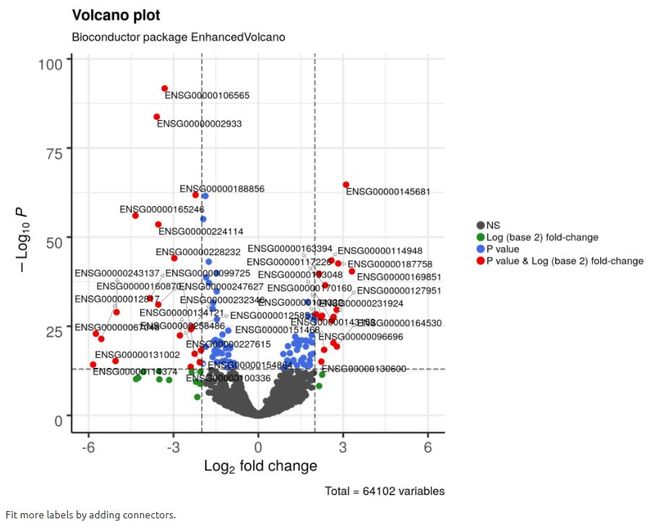
4.8 仅标记关键转录本
在许多情况下,人们可能只希望标记他们感兴趣的关键转录本。 因此,可以通过selectLab参数提要标记的转录本的名字。当然,只有通过差异基因阈值筛选的名字才会被标记。
EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
## 标记目标基因
selectLab = c('ENSG00000106565','ENSG00000187758'),
xlim = c(-6,7),
xlab = bquote(~Log[2]~ 'fold change'),
pCutoff = 10e-14,
FCcutoff = 2.0,
transcriptPointSize = 3.0,
transcriptLabSize = 5.0,
shape = c(4, 35, 17, 18),
colAlpha = 1,
legend=c('NS','Log (base 2) fold-change','P value','P value & Log (base 2) fold-change'),
legendPosition = 'right',
legendLabSize = 14,
legendIconSize = 5.0)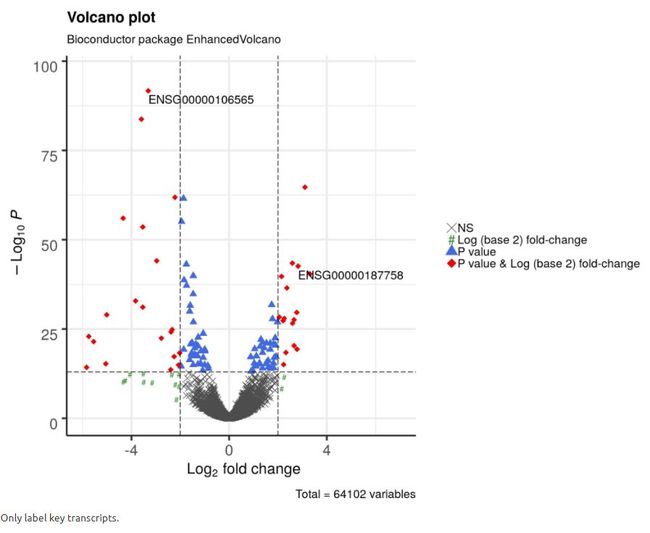
4.9 给标签加框
EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
selectLab = c('ENSG00000106565','ENSG00000187758',
'ENSG00000230795', 'ENSG00000164530',
'ENSG00000143153'),
xlim = c(-5.5,8),
xlab = bquote(~Log[2]~ 'fold change'),
pCutoff = 10e-14,
FCcutoff = 2.0,
transcriptPointSize = 3.0,
transcriptLabSize = 5.0,
transcriptLabCol = 'black',
transcriptLabFace = 'bold',
# 加框
boxedlabels = TRUE,
colAlpha = 4/5,
legend=c('NS','Log (base 2) fold-change','P value','P value & Log (base 2) fold-change'),
legendPosition = 'right',
legendLabSize = 14,
legendIconSize = 4.0,
drawConnectors = TRUE,
widthConnectors = 1.0,
colConnectors = 'black')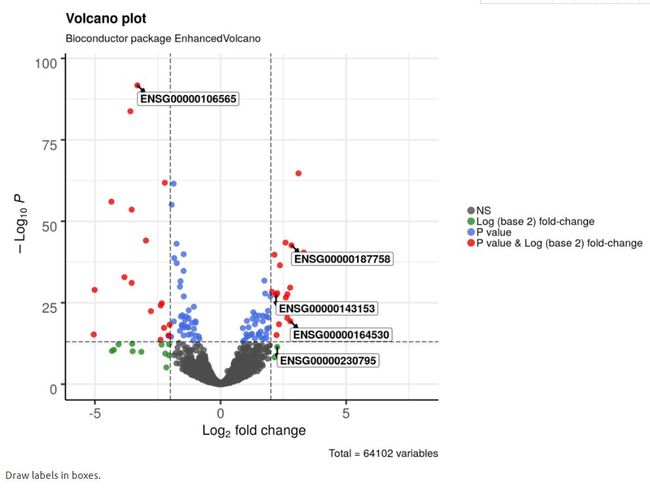
4.10 使用自定义值着色方案
在这个例子,作者希望将log2FC> 2.5的所有转录本标记为“high”,将log2FC <-2.5的转录本标记为“low”。
# create custom key-value pairs for 'high', 'low', 'mid' expression by fold-change
# 通过named vector生成自定义颜色
# set the base colour as 'black'
keyvals <- rep('black', nrow(res2))
# set the base name/label as 'Mid'
names(keyvals) <- rep('Mid', nrow(res2))
# modify keyvals for transcripts with fold change > 2.5
keyvals[which(res2$log2FoldChange > 2.5)] <- 'gold'
names(keyvals)[which(res2$log2FoldChange > 2.5)] <- 'high'
# modify keyvals for transcripts with fold change < -2.5
keyvals[which(res2$log2FoldChange < -2.5)] <- 'royalblue'
names(keyvals)[which(res2$log2FoldChange < -2.5)] <- 'low'
unique(names(keyvals))## [1] "Mid" "low" "high" unique(keyvals)## [1] "black" "royalblue" "gold" keyvals[1:20]## Mid Mid Mid Mid Mid Mid Mid Mid Mid
## "black" "black" "black" "black" "black" "black" "black" "black" "black"
## Mid Mid Mid Mid Mid Mid Mid Mid Mid
## "black" "black" "black" "black" "black" "black" "black" "black" "black"
## Mid Mid
## "black" "black" p1 <- EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
selectLab = rownames(res2)[which(names(keyvals) %in% c('high', 'low'))],
xlim = c(-6.5,6.5),
xlab = bquote(~Log[2]~ 'fold change'),
title = 'Custom colour over-ride',
pCutoff = 10e-14,
FCcutoff = 1.0,
transcriptPointSize = 3.5,
transcriptLabSize = 4.5,
shape = c(6, 4, 2, 11),
# 自定义颜色
colCustom = keyvals,
colAlpha = 1,
legendPosition = 'top',
legendLabSize = 15,
legendIconSize = 5.0,
drawConnectors = TRUE,
widthConnectors = 0.5,
colConnectors = 'grey50',
gridlines.major = TRUE,
gridlines.minor = FALSE,
border = 'partial',
borderWidth = 1.5,
borderColour = 'black')
p2 <- EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
selectLab = rownames(res2)[which(names(keyvals) %in% c('high', 'low'))],
xlim = c(-6.5,6.5),
xlab = bquote(~Log[2]~ 'fold change'),
title = 'No custom colour over-ride',
pCutoff = 10e-14,
FCcutoff = 1.0,
transcriptPointSize = 3.5,
transcriptLabSize = 4.5,
colCustom = NULL,
colAlpha = 1,
legendPosition = 'top',
legendLabSize = 15,
legendIconSize = 5.0,
drawConnectors = FALSE,
widthConnectors = 0.5,
colConnectors = 'grey50',
gridlines.major = TRUE,
gridlines.minor = FALSE,
border = 'full',
borderWidth = 1.0,
borderColour = 'black')
library(gridExtra)
library(grid)
grid.arrange(p1, p2,
ncol=2,
top = textGrob('EnhancedVolcano',
just = c('center'),
gp = gpar(fontsize = 32)))
grid.rect(gp=gpar(fill=NA))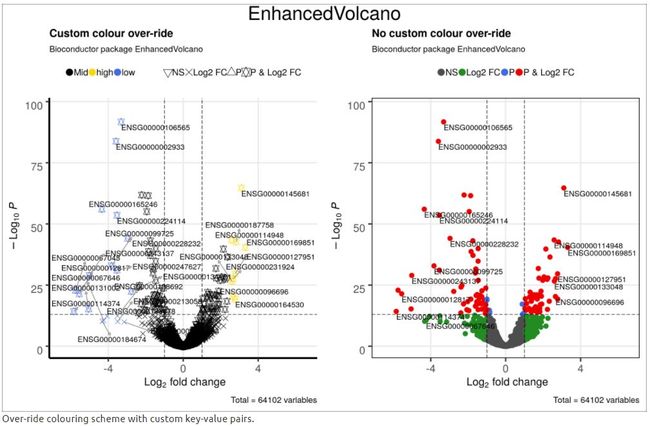
4.11 使用自定义value对覆盖颜色和/或形状进行修改
# define different cell-types that will be shaded
celltype1 <- c('ENSG00000106565', 'ENSG00000002933',
'ENSG00000165246', 'ENSG00000224114')
celltype2 <- c('ENSG00000230795', 'ENSG00000164530',
'ENSG00000143153', 'ENSG00000169851',
'ENSG00000231924', 'ENSG00000145681')
# create custom key-value pairs for different cell-types
# set the base shape as '3'
keyvals.shape <- rep(3, nrow(res2))
# set the base name/label as 'PBC'
names(keyvals.shape) <- rep('PBC', nrow(res2))
# modify the keyvals for cell-type 1
keyvals.shape[which(rownames(res2) %in% celltype1)] <- 17
names(keyvals.shape)[which(rownames(res2) %in% celltype1)] <- 'Cell-type 1'
# modify the keyvals for cell-type 2
keyvals.shape[which(rownames(res2) %in% celltype2)] <- 64
names(keyvals.shape)[which(rownames(res2) %in% celltype2)] <- 'Cell-type 2'
unique(names(keyvals.shape))## [1] "PBC" "Cell-type 1" "Cell-type 2" unique(keyvals.shape)## [1] 3 17 64 keyvals.shape[1:20]## PBC PBC PBC PBC PBC PBC PBC PBC PBC PBC PBC PBC PBC PBC PBC PBC PBC PBC
## 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3 3
## PBC PBC
## 3 3 p1 <- EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
selectLab = rownames(res2)[which(names(keyvals) %in% c('high', 'low'))],
xlim = c(-6.5,6.5),
xlab = bquote(~Log[2]~ 'fold change'),
title = 'Custom shape over-ride',
pCutoff = 10e-14,
FCcutoff = 1.0,
transcriptPointSize = 3.5,
transcriptLabSize = 4.5,
shapeCustom = keyvals.shape,
colCustom = NULL,
colAlpha = 1,
legendLabSize = 15,
legendPosition = 'left',
legendIconSize = 5.0,
drawConnectors = TRUE,
widthConnectors = 0.5,
colConnectors = 'grey50',
gridlines.major = TRUE,
gridlines.minor = FALSE,
border = 'partial',
borderWidth = 1.5,
borderColour = 'black')
# create custom key-value pairs for 'high', 'low', 'mid' expression by fold-change
# set the base colour as 'black'
keyvals.colour <- rep('black', nrow(res2))
# set the base name/label as 'Mid'
names(keyvals.colour) <- rep('Mid', nrow(res2))
# modify keyvals for transcripts with fold change > 2.5
keyvals.colour[which(res2$log2FoldChange > 2.5)] <- 'gold'
names(keyvals.colour)[which(res2$log2FoldChange > 2.5)] <- 'high'
# modify keyvals for transcripts with fold change < -2.5
keyvals.colour[which(res2$log2FoldChange < -2.5)] <- 'royalblue'
names(keyvals.colour)[which(res2$log2FoldChange < -2.5)] <- 'low'
unique(names(keyvals.colour))## [1] "Mid" "low" "high" unique(keyvals.colour)## [1] "black" "royalblue" "gold"p2 <- EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
selectLab = rownames(res2)[which(names(keyvals) %in% c('High', 'Low'))],
xlim = c(-6.5,6.5),
xlab = bquote(~Log[2]~ 'fold change'),
title = 'Custom shape & colour over-ride',
pCutoff = 10e-14,
FCcutoff = 1.0,
transcriptPointSize = 5.5,
transcriptLabSize = 0.0,
shapeCustom = keyvals.shape,
colCustom = keyvals.colour,
colAlpha = 1,
legendPosition = 'top',
legendLabSize = 15,
legendIconSize = 5.0,
drawConnectors = TRUE,
widthConnectors = 0.5,
colConnectors = 'grey50',
gridlines.major = TRUE,
gridlines.minor = FALSE,
border = 'full',
borderWidth = 1.0,
borderColour = 'black')
library(gridExtra)
library(grid)
grid.arrange(p1, p2,
ncol=2,
top = textGrob('EnhancedVolcano',
just = c('center'),
gp = gpar(fontsize = 32)))
grid.rect(gp=gpar(fill=NA))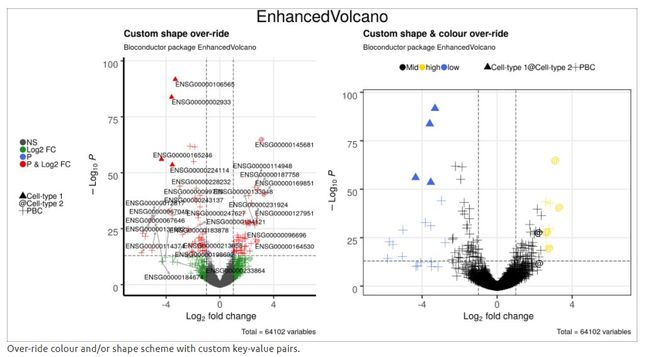
4.12 Shade 指定的转录本
此功能最适用于仅显示1或2个关键转录本。用户可以使用’shapeCustom’参数来更识别不同类型的转录本。
# define different cell-types that will be shaded
celltype1 <- c('ENSG00000106565', 'ENSG00000002933')
celltype2 <- c('ENSG00000230795', 'ENSG00000164530')p1 <- EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
selectLab = celltype1,
xlim = c(-6.5,6.5),
xlab = bquote(~Log[2]~ 'fold change'),
title = 'Shading cell-type 1',
pCutoff = 10e-14,
FCcutoff = 1.0,
transcriptPointSize = 8.0,
transcriptLabSize = 5.0,
transcriptLabCol = 'purple',
transcriptLabFace = 'bold',
boxedlabels = TRUE,
shape = 42,
# 自定义颜色
colCustom = keyvals,
colAlpha = 1,
legendPosition = 'top',
legendLabSize = 15,
legendIconSize = 5.0,
# 自定义标签的背景
shade = celltype1,
shadeLabel = 'Cell-type I',
shadeAlpha = 1/2,
shadeFill = 'purple',
shadeSize = 1,
shadeBins = 5,
drawConnectors = TRUE,
widthConnectors = 1.0,
colConnectors = 'grey30',
gridlines.major = TRUE,
gridlines.minor = FALSE,
border = 'partial',
borderWidth = 1.5,
borderColour = 'black')
p2 <- EnhancedVolcano(res2,
lab = rownames(res2),
x = 'log2FoldChange',
y = 'pvalue',
selectLab = celltype2,
xlim = c(-6.5,6.5),
xlab = bquote(~Log[2]~ 'fold change'),
title = 'Shading cell-type 2',
pCutoff = 10e-14,
FCcutoff = 1.0,
transcriptLabSize = 5.0,
transcriptLabCol = 'forestgreen',
transcriptLabFace = 'bold',
# 自定义形状
shapeCustom = keyvals.shape,
colCustom = keyvals.colour,
colAlpha = 1,
legendPosition = 'top',
transcriptPointSize = 4.0,
legendLabSize = 15,
legendIconSize = 5.0,
shade = celltype2,
shadeLabel = 'Cell-type II',
shadeAlpha = 1/2,
shadeFill = 'forestgreen',
shadeSize = 1,
shadeBins = 5,
drawConnectors = TRUE,
widthConnectors = 1.0,
colConnectors = 'grey30',
gridlines.major = TRUE,
gridlines.minor = FALSE,
border = 'full',
borderWidth = 1.0,
borderColour = 'black')
library(gridExtra)
library(grid)
grid.arrange(p1, p2,
ncol=2,
top = textGrob('EnhancedVolcano',
just = c('center'),
gp = gpar(fontsize = 32)))
grid.rect(gp=gpar(fill=NA))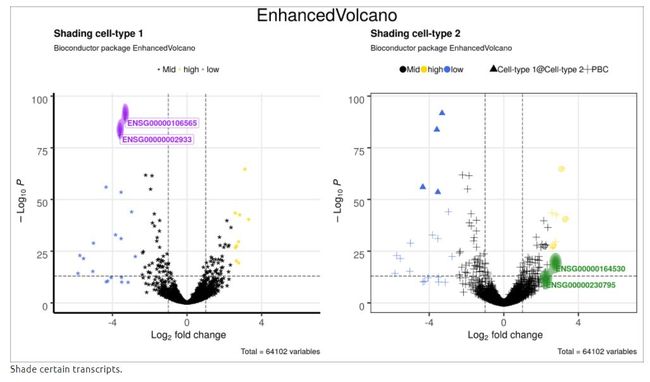
5 Session info
sessionInfo()## R version 3.6.0 (2019-04-26)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 18.04.2 LTS
##
## Matrix products: default
## BLAS: /home/biocbuild/bbs-3.10-bioc/R/lib/libRblas.so
## LAPACK: /home/biocbuild/bbs-3.10-bioc/R/lib/libRlapack.so
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=C
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] grid parallel stats4 stats graphics grDevices utils
## [8] datasets methods base
##
## other attached packages:
## [1] gridExtra_2.3 DESeq2_1.25.0
## [3] magrittr_1.5 airway_1.5.0
## [5] SummarizedExperiment_1.15.1 DelayedArray_0.11.0
## [7] BiocParallel_1.19.0 matrixStats_0.54.0
## [9] Biobase_2.45.0 GenomicRanges_1.37.8
## [11] GenomeInfoDb_1.21.1 IRanges_2.19.6
## [13] S4Vectors_0.23.6 BiocGenerics_0.31.2
## [15] EnhancedVolcano_1.3.1 ggrepel_0.8.1
## [17] ggplot2_3.1.1 knitr_1.23
##
## loaded via a namespace (and not attached):
## [1] bit64_0.9-7 splines_3.6.0 Formula_1.2-3
## [4] assertthat_0.2.1 highr_0.8 latticeExtra_0.6-28
## [7] blob_1.1.1 GenomeInfoDbData_1.2.1 yaml_2.2.0
## [10] RSQLite_2.1.1 pillar_1.4.1 backports_1.1.4
## [13] lattice_0.20-38 glue_1.3.1 digest_0.6.19
## [16] RColorBrewer_1.1-2 XVector_0.25.0 checkmate_1.9.3
## [19] colorspace_1.4-1 htmltools_0.3.6 Matrix_1.2-17
## [22] plyr_1.8.4 XML_3.98-1.19 pkgconfig_2.0.2
## [25] genefilter_1.67.1 zlibbioc_1.31.0 purrr_0.3.2
## [28] xtable_1.8-4 scales_1.0.0 tibble_2.1.2
## [31] htmlTable_1.13.1 annotate_1.63.0 withr_2.1.2
## [34] nnet_7.3-12 lazyeval_0.2.2 survival_2.44-1.1
## [37] crayon_1.3.4 memoise_1.1.0 evaluate_0.14
## [40] MASS_7.3-51.4 foreign_0.8-71 tools_3.6.0
## [43] data.table_1.12.2 stringr_1.4.0 locfit_1.5-9.1
## [46] munsell_0.5.0 cluster_2.0.9 AnnotationDbi_1.47.0
## [49] compiler_3.6.0 rlang_0.3.4 RCurl_1.95-4.12
## [52] rstudioapi_0.10 htmlwidgets_1.3 labeling_0.3
## [55] bitops_1.0-6 base64enc_0.1-3 rmarkdown_1.13
## [58] gtable_0.3.0 DBI_1.0.0 R6_2.4.0
## [61] dplyr_0.8.1 bit_1.1-14 Hmisc_4.2-0
## [64] stringi_1.4.3 Rcpp_1.0.1 geneplotter_1.63.0
## [67] rpart_4.1-15 acepack_1.4.1 tidyselect_0.2.5
## [70] xfun_0.7R统计和作图
文章用图的修改和排版 (1)
文章用图的修改和排版 (2)
简单强大的在线绘图
简单强大的在线绘图-升级版
简单强大的在线绘图-第3版
论文图表基本规范
学术图表的基本配色方法
数据可视化基本套路总结
Graphpad,经典绘图工具初学初探
你的包佩奇了吗?试试新版Rstudio,自动提醒缺失包!
原来Rstudio还可以这么使用,又方便了一些
在R中赞扬下努力工作的你,奖励一份CheatShet
别人的电子书,你的电子书,都在bookdown
R语言 - 入门环境Rstudio
R语言 - 热图绘制 (heatmap)
R语言 - 基础概念和矩阵操作
R语言 - 热图简化
R语言 - 热图美化
R语言 - 线图绘制
R语言 - 线图一步法
R语言 - 箱线图(小提琴图、抖动图、区域散点图)
R语言 - 箱线图一步法
R语言 - 火山图
R语言 - 富集分析泡泡图
R语言 - 散点图绘制
R语言 - 韦恩图
R语言 - 柱状图
R语言 - 图形设置中英字体
R语言 - 非参数法生存分析
R语言 - 绘制seq logo图
WGCNA分析,简单全面的最新教程
psych +igraph:共表达网络构建
一文学会网络分析——Co-occurrence网络图在R中的实现
一文看懂PCA主成分分析
富集分析DotPlot,可以服
基因共表达聚类分析和可视化
R中1010个热图绘制方法
还在用PCA降维?快学学大牛最爱的t-SNE算法吧, 附Python/R代码
一个函数抓取代谢组学权威数据库HMDB的所有表格数据
文章用图的修改和排版
network3D: 交互式桑基图
network3D 交互式网络生成
Seq logo 在线绘制工具——Weblogo
生物AI插图素材获取和拼装指导
ggplot2高效实用指南 (可视化脚本、工具、套路、配色)
图像处理R包magick学习笔记
SOM基因表达聚类分析初探
利用gganimate可视化全球范围R-Ladies(R社区性别多样性组织)发展情况
一分钟绘制磷脂双分子层:AI零基础入门和基本图形绘制
AI科研绘图(二):模式图的基本画法
你知道R中的赋值符号箭头(<-)和等号(=)的区别吗?
R语言可视化学习笔记之ggridges包
利用ComplexHeatmap绘制热图(一)
ggplot2学习笔记之图形排列
R包reshape2,轻松实现长、宽数据表格转换
用R在地图上绘制网络图的三种方法
PCA主成分分析实战和可视化 附R代码和测试数据
iTOL快速绘制颜值最高的进化树!
12个ggplot2扩展包帮你实现更强大的可视化
编程模板-R语言脚本写作:最简单的统计与绘图,包安装、命令行参数解析、文件读取、表格和矢量图输出
R语言统计入门课程推荐——生物科学中的数据分析Data Analysis for the Life Sciences
数据可视化基本套路总结
你知道R中的赋值符号箭头
<-和等号=的区别吗?使用dplyr进行数据操作30例
交集intersect、并集union、找不同setdiff
R包reshape2,轻松实现长、宽数据表格转换
1数据类型(向量、数组、矩阵、 列表和数据框)
2读写数据所需的主要函数、与外部环境交互
3数据筛选——提取对象的子集
4向量、矩阵的数学运算
5控制结构
6函数及作用域
7认识循环函数lapply和sapply
8分解数据框split和查看对象str
9模拟—随机数、抽样、线性模型
1初识ggplot2绘制几何对象
2图层的使用—基础、加标签、注释
3工具箱—误差线、加权数、展示数据分布
4语法基础
5通过图层构建图像
6标度、轴和图例
7定位-分面和坐标系
8主题设置、存储导出
9绘图需要的数据整理技术
创建属于自己的调色板
28个实用绘图包,总有几个适合你
热图绘制
R做线性回归
绘图相关系数矩阵corrplot
相关矩阵可视化ggcorrplot
绘制交互式图形recharts
交互式可视化CanvasXpress
聚类分析factoextra
LDA分析、作图及添加置信-ggord
解决散点图样品标签重叠ggrepel
添加P值或显著性标记ggpubr
Alpha多样性稀释曲线rarefraction curve
堆叠柱状图各成分连线画法:突出组间变化
冲击图展示组间时间序列变化ggalluvial
桑基图riverplot
微生物环境因子分析ggvegan
五彩进化树与热图更配ggtree
多元回归树分析mvpart
随机森林randomForest 分类Classification 回归Regression
加权基因共表达网络分析WGCNA
circlize包绘制circos-plot
R语言搭建炫酷的线上博客系统
维恩(Venn)图绘制工具大全 (在线+R包)
R包circlize:柱状图用腻了?试试好看的弦状图
易生信系列培训课程,扫码获取免费资料

更多阅读
画图三字经 生信视频 生信系列教程
心得体会 TCGA数据库 Linux Python
高通量分析 免费在线画图 测序历史 超级增强子
生信学习视频 PPT EXCEL 文章写作 ggplot2
海哥组学 可视化套路 基因组浏览器
色彩搭配 图形排版 互作网络
自学生信 2019影响因子 GSEA 单细胞
后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集

