SCS【6】单细胞转录组之细胞类型自动注释 (SingleR)

点击关注,桓峰基因
桓峰基因公众号推出单细胞系列教程,有需要生信分析的老师可以联系我们!首选看下转录分析教程整理如下:
Topic 6. 克隆进化之 Canopy
Topic 7. 克隆进化之 Cardelino
Topic 8. 克隆进化之 RobustClone
SCS【1】今天开启单细胞之旅,述说单细胞测序的前世今生
SCS【2】单细胞转录组 之 cellranger
SCS【3】单细胞转录组数据 GEO 下载及读取
SCS【4】单细胞转录组数据可视化分析 (Seurat 4.0)
SCS【5】单细胞转录组数据可视化分析 (scater)
SCS【6】单细胞转录组之细胞类型自动注释 (SingleR)
今天来说说单细胞转录组数据的细胞类型的注释,学会这些分析结果,距离发文章就只差样本的选择了,有创新性的样本将成为文章的亮点,并不是分析内容了!
前 言
单细胞研究中细胞类型注释是很重要的环节,大致分为人工注释和软件注释。
(1)人工注释
人工注释需要借助文献检索marker或者结合常用的注释数据库:
cellMarker,PanglaoDB, CancerSEA等,比较考验研究者的相关背景和精力,优点在于准确性相对较好。
(2)软件自动化注释
软件自动化注释一般是使用软件内置数据集进行注释,操作相对简单。但是准确性会相对稍差,不过可以作为一种很好的辅助注释手段。
自动化注释的软件很多,本次先简单分享如何使用singleR进行自动注释。
SingleR是一个用于对单细胞RNA-seq测序(scRNA-seq)数据进行细胞类型自动注释的R包(Aran et al.2019)。依据已知类型标签的细胞样本作为参考数据集,对测试数据集中的细胞进行标记注释。
SingleR是一种用于单细胞RNA测序(scRNAseq)数据的自动标注方法(Aran et al. 2019)。给定一个具有已知标签的参考样本集(单细胞或批量),它根据与参考的相似性对来自测试数据集的新单元格进行标记。因此,对于参考数据集,手动解释集群和定义标记基因的负担只需要做一次,并且这种生物知识可以以自动的方式传播到新的数据集。singleR自带的7个参考数据集,其中5个是人类数据,2个是小鼠的数据:
1. BlueprintEncodeData Blueprint (Martens and Stunnenberg 2013) and Encode (The ENCODE Project Consortium 2012) (人)
2. DatabaseImmuneCellExpressionData The Database for Immune Cell Expression(/eQTLs/Epigenomics)(Schmiedel et al. 2018)(人)
3. HumanPrimaryCellAtlasData the Human Primary Cell Atlas (Mabbott et al. 2013)(人)
4. MonacoImmuneData, Monaco Immune Cell Data - GSE107011 (Monaco et al. 2019)(人)
5. NovershternHematopoieticData Novershtern Hematopoietic Cell Data - GSE24759(人)
ImmGenData the murine ImmGen (Heng et al. 2008) (鼠)
MouseRNAseqData a collection of mouse data sets downloaded from GEO (Benayoun et al. 2019).鼠)
为了简明扼要,本文只简要介绍了SingleR的基本功能。然而,该包还提供了更高级的功能,包括同时使用多个引用,操作细胞本体和提高大数据集的性能。读者可以参考这本书了解更多细节。
软件安装
if(!require(celldex))
BiocManager::install("celldex")
if(!require(SingleR))
BiocManager::install("SingleR")
数据读取
这里我们同样使用 SCS【4】单细胞转录组数据可视化分析 (Seurat 4.0) 数据例子,具体数据的处理过程这里都有,直接运行即可,此外关于降维聚类我们三种方法都有分析,三个不同是函数处理降维:RunPCA(), RunUMAP(), RunTSNE()。当我们输出pbmc时,可以看到 下面一行信息,“3 dimensional reductions calculated: pca, umap, tsne”,说明三种降维方法都已完成。
library(Seurat)
# 进行singleR注释
pbmc.data <- Read10X(data.dir = "./filtered_gene_bc_matrices/hg19")
# Initialize the Seurat object with the raw (non-normalized data).
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3,
min.features = 200)
pbmc
## An object of class Seurat
## 13714 features across 2700 samples within 1 assay
## Active assay: RNA (13714 features, 0 variable features)
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
pbmc <- NormalizeData(pbmc)
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, features = all.genes)
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))
pbmc <- FindNeighbors(pbmc, dims = 1:10)
pbmc <- FindClusters(pbmc, resolution = 0.5)
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 2700
## Number of edges: 97892
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.8719
## Number of communities: 9
## Elapsed time: 0 seconds
pbmc <- RunUMAP(pbmc, dims = 1:10)
pbmc <- RunTSNE(pbmc, dims = 1:10)
pbmc
## An object of class Seurat
## 13714 features across 2700 samples within 1 assay
## Active assay: RNA (13714 features, 2000 variable features)
## 3 dimensional reductions calculated: pca, umap, tsne
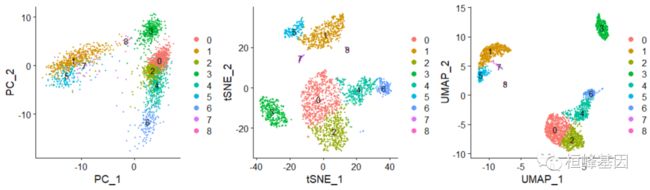
绘制三种不同降维聚类的结果,如下:
plot1 <- DimPlot(pbmc, reduction = "pca", label = TRUE)
plot2 <- DimPlot(pbmc, reduction = "tsne", label = TRUE)
plot3 <- DimPlot(pbmc, reduction = "umap", label = TRUE)
plot1 + plot2 + plot3
数据库加载
我们选取两个 human相关的单细胞类型数据库,我们加载之后可以将变量保存起来,以免下次使用比较耗时,保存之后直接load即可,如下:
library(SingleR)
library(celldex)
### 数据库加载 hpca.se <- celldex::HumanPrimaryCellAtlasData() hpca.se@metadata
### bpe.se<-celldex::BlueprintEncodeData() bpe.se@metadata
### save(hpca.se,file='HumanPrimaryCellAtlas_hpca.se_human.RData')
### save(bpe.se,file='BlueprintEncode_bpe.se_human.RData')
load("HumanPrimaryCellAtlas_hpca.se_human.RData")
load("BlueprintEncode_bpe.se_human.RData")
单个数据库注释
只选择单个数据库,类似我们的细胞具有哪一类细胞的倾向性。
pbmc4SingleR <- GetAssayData(pbmc, slot = "data") ##获取标准化矩阵
pbmc.hesc <- SingleR(test = pbmc4SingleR, ref = hpca.se, labels = hpca.se$label.main) #
pbmc.hesc
## DataFrame with 2700 rows and 5 columns
## scores first.labels
##
## AAACATACAACCAC-1 0.1031269:0.234280:0.222822:... T_cells
## AAACATTGAGCTAC-1 0.0984977:0.361135:0.300626:... B_cell
## AAACATTGATCAGC-1 0.0671635:0.266281:0.237642:... T_cells
## AAACCGTGCTTCCG-1 0.0836640:0.235213:0.273059:... Monocyte
## AAACCGTGTATGCG-1 0.0739783:0.164291:0.175458:... NK_cell
## ... ... ...
## TTTCGAACTCTCAT-1 0.0965528:0.249749:0.307289:... Pre-B_cell_CD34-
## TTTCTACTGAGGCA-1 0.1368350:0.302956:0.269191:... Pre-B_cell_CD34-
## TTTCTACTTCCTCG-1 0.0749830:0.272021:0.225157:... B_cell
## TTTGCATGAGAGGC-1 0.0673739:0.232823:0.184013:... B_cell
## TTTGCATGCCTCAC-1 0.0890275:0.242453:0.243141:... T_cells
## tuning.scores labels pruned.labels
##
## AAACATACAACCAC-1 0.301975:0.2133145 T_cells T_cells
## AAACATTGAGCTAC-1 0.326636:0.0220241 B_cell B_cell
## AAACATTGATCAGC-1 0.307280:0.1090948 T_cells T_cells
## AAACCGTGCTTCCG-1 0.286386:0.2360274 Monocyte Monocyte
## AAACCGTGTATGCG-1 0.299925:0.2107053 NK_cell NK_cell
## ... ... ... ...
## TTTCGAACTCTCAT-1 0.259345:0.134353 Monocyte Monocyte
## TTTCTACTGAGGCA-1 0.157336:0.129647 Pre-B_cell_CD34- Pre-B_cell_CD34-
## TTTCTACTTCCTCG-1 0.235417:0.173613 B_cell B_cell
## TTTGCATGAGAGGC-1 0.228548:0.149524 B_cell B_cell
## TTTGCATGCCTCAC-1 0.288418:0.194118 T_cells T_cells
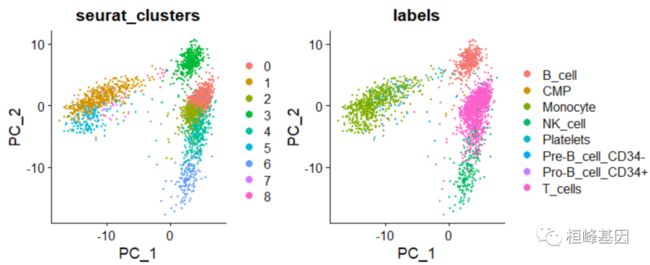
绘制降维聚类图
PCA 降维聚类
# seurat 和 singleR的table表
table(pbmc.hesc$labels, pbmc$seurat_clusters)
##
## 0 1 2 3 4 5 6 7 8
## B_cell 0 0 0 339 0 0 2 0 0
## CMP 2 0 2 0 0 0 0 0 0
## Monocyte 0 444 0 0 0 152 0 31 2
## NK_cell 4 0 1 0 54 0 139 0 0
## Platelets 0 0 0 0 0 0 0 0 12
## Pre-B_cell_CD34- 4 47 1 1 1 6 2 4 0
## Pro-B_cell_CD34+ 0 0 0 4 0 0 0 0 1
## T_cells 686 0 463 5 284 1 5 1 0
[email protected]$labels <- pbmc.hesc$labels
DimPlot(pbmc, group.by = c("seurat_clusters", "labels"), reduction = "pca")
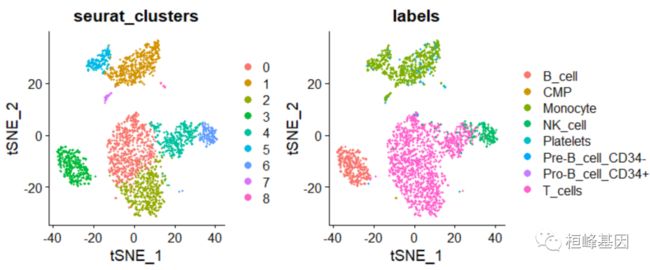
t-SNE 降维聚类
DimPlot(pbmc, group.by = c("seurat_clusters", "labels"),reduction = "tsne")
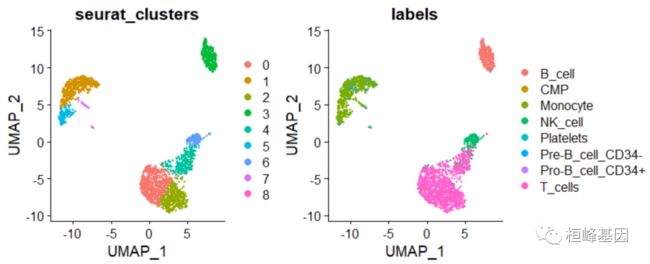
UMAP 降维聚类
DimPlot(pbmc, group.by = c("seurat_clusters", "labels"),reduction = "umap")
多个数据库注释
使用BP和HPCA两个数据库综合注释,使用list函数读入多个数据库。
PCA 降维聚类
pbmc.hesc <- SingleR(test = pbmc4SingleR, ref = list(BP = bpe.se, HPCA = hpca.se),
labels = list(bpe.se$label.main, hpca.se$label.main))
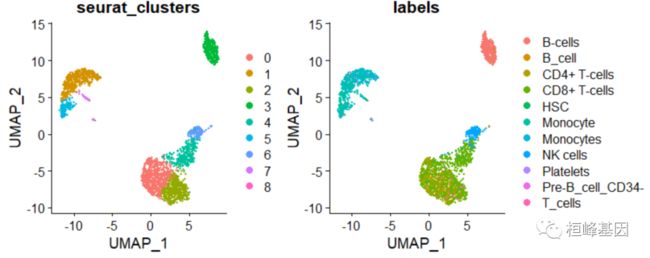
table(pbmc.hesc$labels, pbmc$seurat_clusters)
##
## 0 1 2 3 4 5 6 7 8
## B-cells 0 0 0 340 0 0 2 1 1
## B_cell 0 0 0 4 0 0 1 0 0
## CD4+ T-cells 298 0 159 1 1 0 0 0 0
## CD8+ T-cells 376 0 282 3 294 0 7 1 0
## HSC 2 0 2 0 0 0 0 1 0
## Monocyte 0 297 0 0 0 125 0 25 1
## Monocytes 0 181 0 0 0 32 0 8 1
## NK cells 2 0 0 0 44 0 138 0 0
## Platelets 0 0 0 0 0 0 0 0 12
## Pre-B_cell_CD34- 0 13 0 0 0 1 0 0 0
## T_cells 18 0 24 1 0 1 0 0 0
[email protected]$labels <- pbmc.hesc$labels
DimPlot(pbmc, group.by = c("seurat_clusters", "labels"), reduction = "pca")

t-SNE 降维聚类
DimPlot(pbmc, group.by = c("seurat_clusters", "labels"),reduction = "tsne")
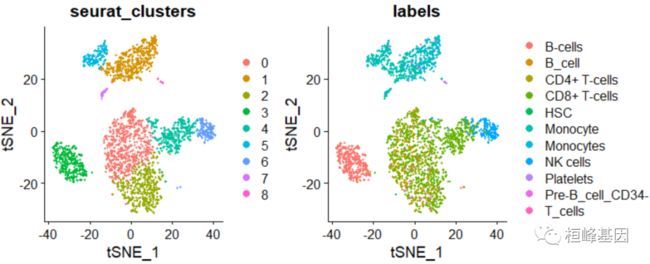
UMAP 降维聚类
DimPlot(pbmc, group.by = c("seurat_clusters", "labels"),reduction = "umap")
注释结果诊断
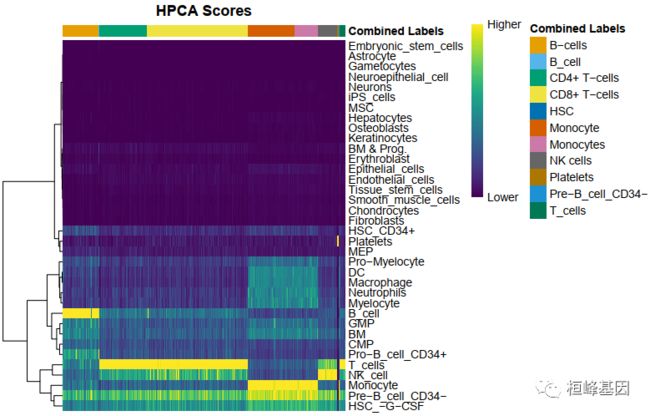
1. 基于scores within cells
细胞在一个标签的得分很显著的高于其他标签得分,注释结果比较清晰。
### Annotation diagnostics
plotScoreHeatmap(pbmc.hesc)
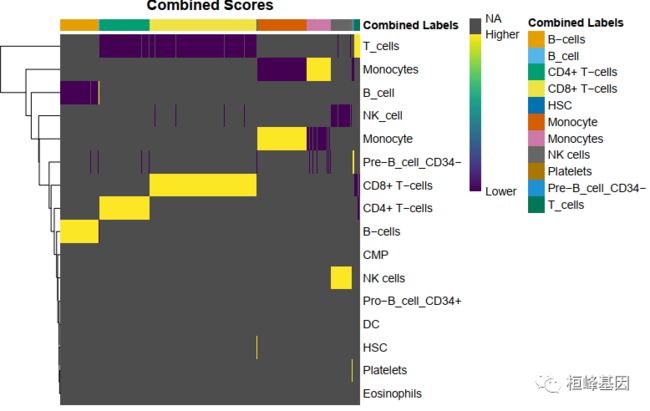
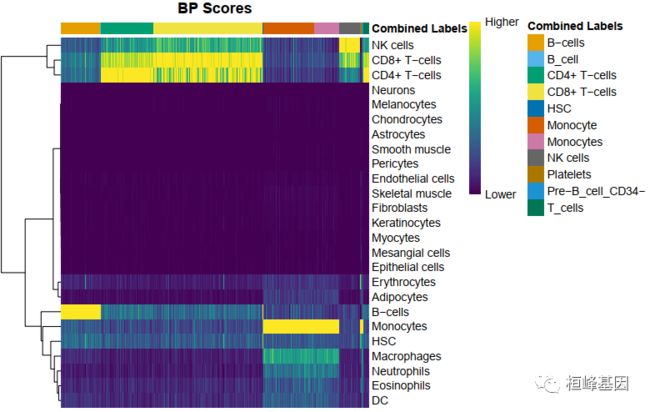
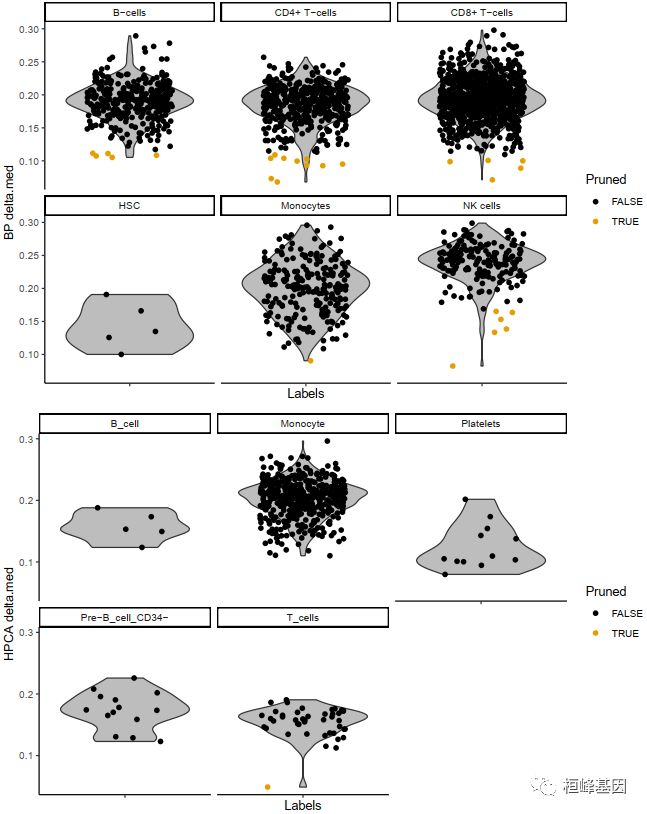
2. 基于 per-cell “deltas”诊断
Delta值低,说明注释结果不是很明确。
plotDeltaDistribution(pbmc.hesc, ncol = 3)
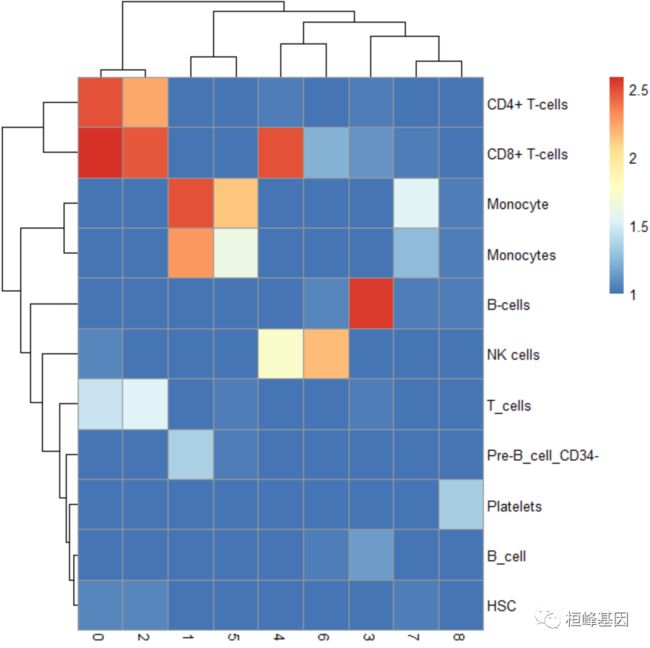
3. 与cluster结果比较
tab <- table(label = pbmc.hesc$labels, cluster = pbmc$seurat_clusters)
tab
## cluster
## label 0 1 2 3 4 5 6 7 8
## B-cells 0 0 0 340 0 0 2 1 1
## B_cell 0 0 0 4 0 0 1 0 0
## CD4+ T-cells 298 0 159 1 1 0 0 0 0
## CD8+ T-cells 376 0 282 3 294 0 7 1 0
## HSC 2 0 2 0 0 0 0 1 0
## Monocyte 0 297 0 0 0 125 0 25 1
## Monocytes 0 181 0 0 0 32 0 8 1
## NK cells 2 0 0 0 44 0 138 0 0
## Platelets 0 0 0 0 0 0 0 0 12
## Pre-B_cell_CD34- 0 13 0 0 0 1 0 0 0
## T_cells 18 0 24 1 0 1 0 0 0
library(pheatmap)
pheatmap(log10(tab + 10))
所有的可视化分析就基本完成了,学会了单细胞分析就非常简单了,目前测序的费用也在降低,单细胞系列可算是目前的测序神器,有这方面需求的老师,联系桓峰基因,提供最高端的科研服务!
桓峰基因,铸造成功的您!
未来桓峰基因公众号将不间断的推出单细胞系列生信分析教程,
敬请期待!!
有想进生信交流群的老师可以扫最后一个二维码加微信,备注“单位+姓名+目的”,有些想发广告的就免打扰吧,还得费力气把你踢出去!
References:
-
Aran, D., A. P. Looney, L. Liu, E. Wu, V. Fong, A. Hsu, S. Chak, et al. 2019. “Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage.” Nat. Immunol. 20 (2): 163–72.
-
Grun, D., M. J. Muraro, J. C. Boisset, K. Wiebrands, A. Lyubimova, G. Dharmadhikari, M. van den Born, et al. 2016. “De Novo Prediction of Stem Cell Identity using Single-Cell Transcriptome Data.” Cell Stem Cell 19 (2): 266–77.
-
La Manno, G., D. Gyllborg, S. Codeluppi, K. Nishimura, C. Salto, A. Zeisel, L. E. Borm, et al. 2016. “Molecular Diversity of Midbrain Development in Mouse, Human, and Stem Cells.” Cell 167 (2): 566–80.
-
Mabbott, Neil A., J. K. Baillie, Helen Brown, Tom C. Freeman, and David A. Hume. 2013. “An expression atlas of human primary cells: Inference of gene function from coexpression networks.” BMC Genomics 14. https://doi.org/10.1186/1471-2164-14-632.
-
Muraro, M. J., G. Dharmadhikari, D. Grun, N. Groen, T. Dielen, E. Jansen, L. van Gurp, et al. 2016. “A Single-Cell Transcriptome Atlas of the Human Pancreas.” Cell Syst 3 (4): 385–94.