Chapter9 : De Novo Molecular Design with Chemical Language Models
reading notes of《Artificial Intelligence in Drug Design》
文章目录
- 1.Introduction
- 2.Materials
-
- 2.1.Computational Methods
- 2.2.Data
- 3.Methods
-
- 3.1.SMILES Notation
- 3.2.Recurrent Neural Networks
-
- 3.2.1.Recurrent Neural Networks
- 3.2.2.BIMODAL
- 3.2.3.One-Hot Encoding
- 3.2.4.Transfer Learning
- 3.3.Training and Sampling Settings
-
- 3.3.1.Model Type
- 3.3.2.Network Architecture and Size
- 3.3.3.Starting Point Positioning
- 3.3.4.Augmentation
- 3.3.5.Number of Fine-Tuning Epochs
- 3.3.6.Sampling Temperature
- 3.3.7.Number of SMILES to Sample
- 3.4.Generating Focused Molecule Libraries
-
- 3.4.1.Molecule Preparation
- 3.4.2.Model Pretraining
- 3.4.3.Fine-Tuning and Sampling
- 3.5.Analysis of the Results
-
- 3.5.1.Cross-Entropy Loss
- 3.5.2.Number of Valid, Unique, and Novel SMILES Strings
- 3.5.3.Scaffold Diversity
- 3.5.4.Structural Similarity of De Novo Designs and Fine-Tuning Molecules
- 3.5.5.Selection of De Novo Design
- 3.6.Final Considerations
1.Introduction
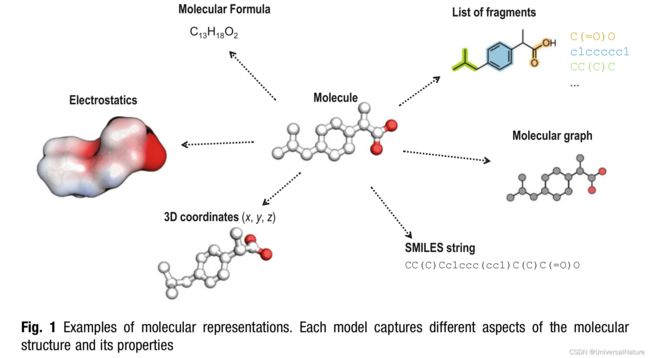
- These molecular representations are human-made models designed to capture certain properties. The molecules possess the syntactic properties and semantic properties.
- Several factors contribute to the popularity of SMILES in the context of deep learning:
- SMILES are strings, which renders them suitable as inputs to sequence modeling algorithms.
- Compared to other string-based molecular representations such as InChI, SMILES strings have a straightforward syntax. This permissive syntax allows for a certain “flexibility of expression”.
- SMILES are easily legible and interpretable by humans.
- The tool example demonstrates how deep learning methods can be employed to generate sets of new SMILES strings, inspired by the structures of four known retinoid X receptor (RXR) modulators, using a recently developed method, the bidirectional molecule generation with alternate learning (BIMODAL). The program code is freely available here.
2.Materials
2.1.Computational Methods
- All calculations were performed using Python 3.7.4 in Jupyter Notebooks. The models rely on PyTorch and RDKit.
- After installing Anaconda and Git, we can run the code below:
git clone https://github.com/ETHmodlab/de_novo_design_RNN.git
cd <path\to\folder>
conda env crate -f environment.yml
conda activate de_novo
cd example
jupyter notebook
2.2.Data
- To emulate a realistic scenario, we provide a tool molecule library containing four RXR modulators (Fig. 2). Molecule 1 is bexarotene, a pharmacological RXR agonist. Molecules 2–4 were obtained from ChEMBL and have a potency on RXR (expressed as EC50, IC50, Ki, or Kd) below0.8μM. This set of bioactive compounds (available in the repository, under “/exam- ple/fine_tuning.csv”) will be used to generate a focused library of de novo designs.

3.Methods
3.1.SMILES Notation
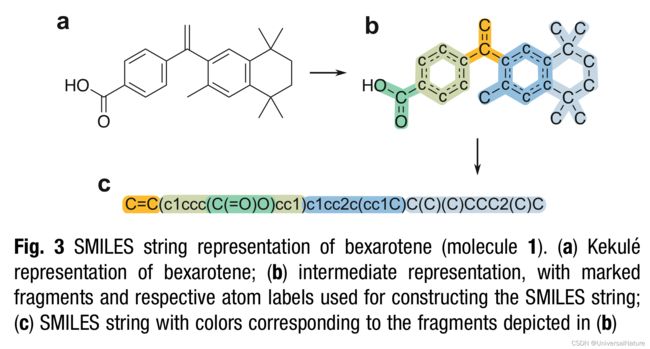
- Stereochemical information is not mandatory but can be specified. The configuration of double bonds is specified using the characters “/” and “\” to indicate directional single bonds adja- cent to a double bond; the configuration of tetrahedral carbons is specified by “@” or “@@”.

3.2.Recurrent Neural Networks
3.2.1.Recurrent Neural Networks
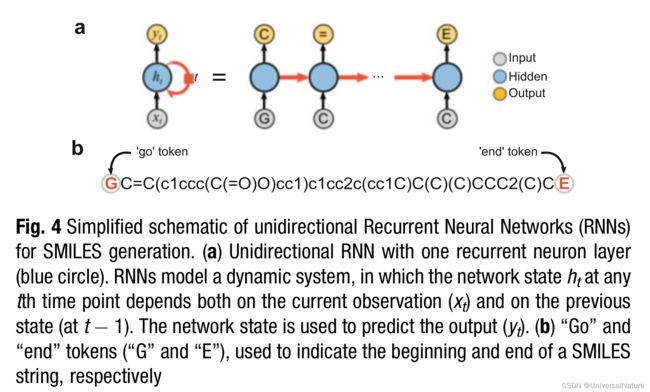
- To generate new SMILES strings, the start token G can be used as the first input character; the RNN model will then extend this one-token string by attaching valid SMILES characters sequentially, until the end token is output.
- While “vanilla” RNNs can, in principle, handle sequences of any length, they are in practice challenged by long-term dependencies, which can lead to gradient vanishing issues during network training. To overcome this limitation, alternative architectures have been proposed, the most popular of which are long short-term memory (LSTM).
3.2.2.BIMODAL
- The BIMODAL approach is an RNN-based method that was specifically designed for SMILES string generation.
- This non-univocity and non-directionality motivated the development of the BIMODAL sequence generation method, which reads and generates SMILES strings both in the forward and backward directions.

- Similar to bidirectional RNNs for supervised learning, BIMODAL consists of two RNNs, each for reading the sequence in one direction. The information captured by each RNN is then combined to provide a joint prediction.
- The effect of “G” token positioning on SMILES string generation is discussed in Subheading 3.3.
3.2.3.One-Hot Encoding

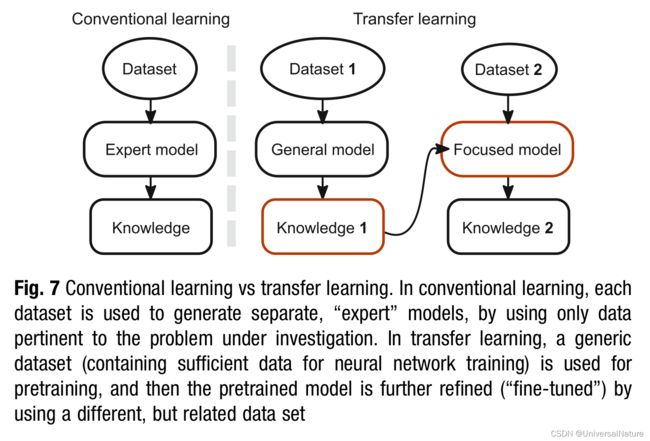
3.2.4.Transfer Learning
3.3.Training and Sampling Settings

3.3.1.Model Type
- The code repository contains two types of models, namely the BIMODAL and the classical “forward” RNNs. The BIMODAL method was used for the worked example. Users can adapt the computational pipeline to forward RNNs, as explained in the accompanying Jupyter notebook.
3.3.2.Network Architecture and Size
- The published BIMODAL architecture is based on two levels of information processing. Each processing level is characterized by two LSTM layers (one for forward and one for backward processing), whose information is combined to predict the next token (Subheading 3.3.3).
3.3.3.Starting Point Positioning

3.3.4.Augmentation
- The possibility of placing the start token at any arbitrary position of the string (i.e., random starting position) allows the performance of a novel type of data augmentation that was introduced for BIMODAL. For each training molecule, one can generate n repetitions of the same SMILES string, in which the start token is placed in a different random position.
3.3.5.Number of Fine-Tuning Epochs
- The choice of the number of fine-tuning epochs is usually made on a case-by-case basis by considering (a) the total training time, which is the product of the training time per epoch and the number of epochs, (b) the desired structural diversity of the de novo designs, which generally decreases with increasing number of fine-tuning epochs, and © the desired similarity of the de novo designs and the fine-tuning set in terms of physicochemical properties, which generally increase with longer fine-tuning.
3.3.6.Sampling Temperature
- Trained language models can be used as generative methods for sampling novel SMILES strings. One possible approach is temperature sampling. By setting the temperature (T), one can govern the randomness of the generated sequences: q i = e z i / T ∑ e z i / T q_i=\frac{e^{z_i/T}}{\sum e^{z_i/T}} qi=∑ezi/Tezi/T, where zi is the RNN prediction for the ith token, j runs over all the tokens in the dictionary, and qi is the probability of sampling the ith token.
- In other words, SMILES are sampled using a Softmax function that is controlled by parameter T. For low values of T, the most likely token according to the estimated probability distribution is selected. With increasing values of T, the probability of selecting the most likely token decreases, and the model generates more diverse sequences (Fig. 10).
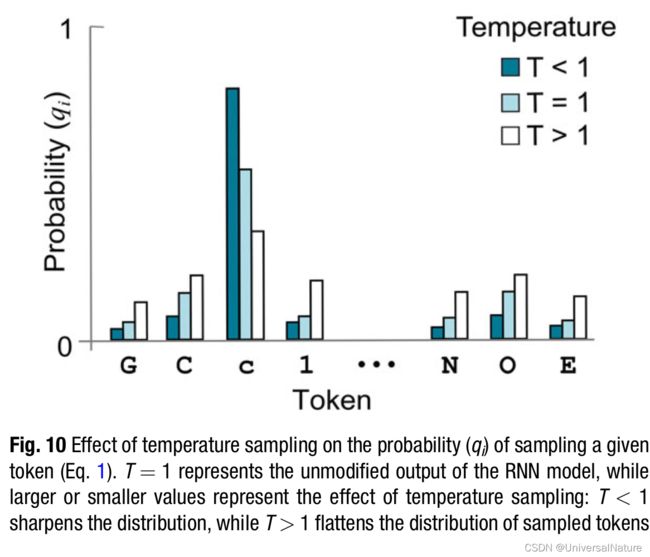
3.3.7.Number of SMILES to Sample
- The higher the number of SMILES generated using temperature sampling, the higher the possibility to explore the chemical space. Here, we sampled 1000 SMILES strings for each fine-tuning epoch, as described below.
3.4.Generating Focused Molecule Libraries
3.4.1.Molecule Preparation
- The pretraining data and fine-tuning set were prepared according to the following procedure:
- Removal invalid, duplicate salt, stereochemical SMILES strings. Note that the SMILES token denoting disconnected structures (“.”) was not present in the BIMODAL token dictionary. Thus, it is not possible to use molecules containing this symbol for training, e.g., SMILES strings representing salt forms.
- SMILES string canonicalization. In this work, canonical SMILES were used for two main reasons: (a) consistency with the original study, and (b) availability of the BIMODAL augmentation strategy for the “G” token position, which allows to generate a sufficient data volume without the need for additional data augmentation.
- Removal of SMILES strings with out-of-bound dimensions. In our data pretreatment pipeline, only SMILES strings encompassing 34–74 tokens were retained.
- Addition of start and end token and data augmentation.
- SMILES string padding.

3.4.2.Model Pretraining
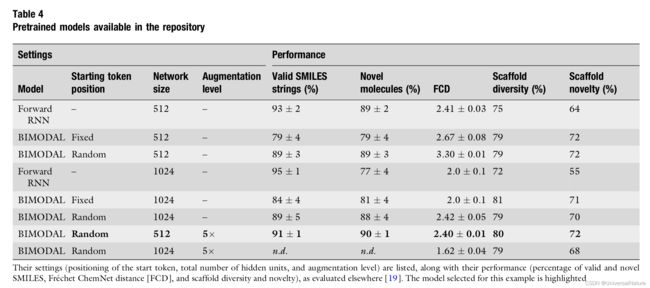
Table 4 contains the results of an evaluation of each of the eight models for the following criteria:
- Percentage of valid and novel molecules.
- Frechet ChemNet Distance (FCD). Generative models should be able to sample molecules with the desired chemical and biological properties. This aspect was evaluated by computing the Frechet ChemNet distance (FCD). The FCD values are based on the activation of the penultimate layer of an LSTM model, which was trained to predict bioactivity. The lower the FCD between two sets of molecules, the closer they are in terms of their structural and predicted biological properties.
- Scaffold diversity and scaffold novelty.
3.4.3.Fine-Tuning and Sampling
- Transfer learning is performed by updating a pretrained network with fine-tuned data.
3.5.Analysis of the Results
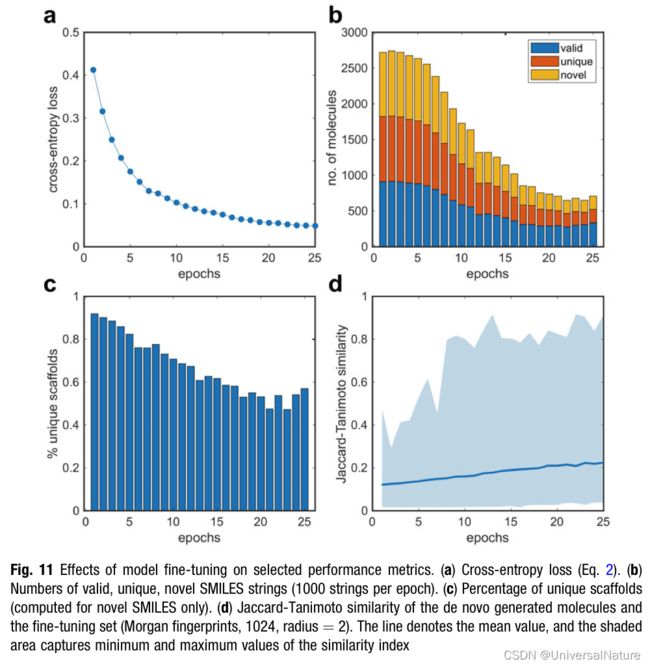
3.5.1.Cross-Entropy Loss
- The cross-entropy loss (L) is computed during model training: L = − ∑ t l o g P ( x ^ t + 1 ∣ x 1 , . . . , x t ) L=-\sum_t logP(\hat{x}_{t+1}|x_1,...,x_t) L=−∑tlogP(x^t+1∣x1,...,xt), where x ^ t + 1 \hat x_{t+1} x^t+1 represents the correct token at position t+1. The cross-entropy loss captures the capacity of the language model to predict the correct token at the step t + 1, given the sequence of preceding tokens.
3.5.2.Number of Valid, Unique, and Novel SMILES Strings
- Validity, uniqueness, and novelty are measures of a model’s capacity to (a) learn the syntax of the chemical language (“validity”), (b) capture its capacity to explore the chemical space (“unique- ness”), and © explore uncharted regions of the chemical space defined by the training data (“novelty”).
3.5.3.Scaffold Diversity
- Identifying isofunctional molecular structures with different scaffolds is a central goal of de novo drug design. A criterion for evaluating generative methods is their ability to construct molecules with diverse chemical scaffolds. The fraction of unique atomic scaffolds of the total number of novel molecules generated is shown in Fig. 11c.
3.5.4.Structural Similarity of De Novo Designs and Fine-Tuning Molecules
- One way to compute the similarity between two molecules in terms of their fragments/functional groups is to use Morgan fingerprints (also known as circular or extended connectivity fingerprints), which are among the most popular molecular descriptors in chemoinformatics. Morgan fingerprints can be used to compute the chemical similarity between two molecules (X and Y), for example, by using the Jaccard–Tanimoto similarity index: S = a a + b S=\frac{a}{a+b} S=a+ba
where a represents the number of bits equal to 1 in the fingerprints of both molecules, and b is the number of set bits differing between the fingerprints of X and Y (i.e., equal to 0 for one fingerprint and 1 for the other). - The Tanimoto index (S) captures the number of bits that are equal to 1 in both fingerprints, over the total number of bits being equal to 1 in at least one of the fingerprint vectors. A greater value of the similarity index (S) suggests greater structural similarity between X and Y. The average Morgan fingerprint similarity decreases with increasing fine-tuning epochs (Fig. 11d) as a result of the model convergence toward the chemical space of the RXR modulators used for fine-tuning.
3.5.5.Selection of De Novo Design
- Epoch 10 is, in fact, characterized by optimal values of all criteria analyzed, e.g., a low loss (L 1⁄4 0.10), 57% novel compounds among the generated SMILES strings, which also have 71% diverse scaffolds, and a wide span of Jaccard–Tanimoto similarity (Morgan fingerprints) ranging from 0.017 to 0.81. Further
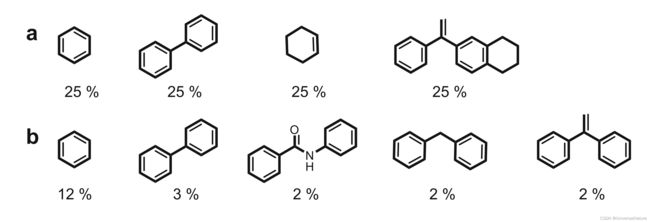
more, the designs generated in epoch 10 possess several novel scaffolds compared to the fine-tuning molecules (Fig. 12)
3.6.Final Considerations
- When applying chemical language models to de novo design, it is important to bear in mind that there is no one-fit-all approach. The approach chosen to navigate and prioritize virtual libraries is possibly the most critical factor in determining the success of prospective applications. A retrospective analysis of the usefulness of the chosen deep learning framework and the validity of the prioritization protocol is recommended.
- Aiming to improve the capabilities of chemical language models, several “hybrid” approaches have been designed, which consider additional aspects for molecule generation, e.g., predicted bioactivity , three-dimensional shape of molecules, and gene signatures. Such approaches will propel chemical language models for medicinal chemistry.