论文解读:基于图神经网络与蛋白质接触图的药靶亲和力预测(一)2021SC@SDUSC
2021SC@SDUSC
论文解读:基于图神经网络与蛋白质接触图的药靶亲和力预测(一)
Drug–target affinity prediction using graph neural
network and contact maps
摘要
计算机辅助药物设计是利用高性能计算机模拟药物设计任务的一个很有前景的研究领域。药物靶标亲和度(DTA)预测是计算机辅助药物设计中最重要的一步,可以加快药物开发速度,减少资源消耗。随着深度学习的发展,将深度学习引入到DTA预测中,提高DTA预测的准确性已成为研究的热点。本文利用分子和蛋白质的结构信息,分别建立了药物分子和蛋白质的图。
introduction
一些背景的介绍引入:
计算机性能背景在深度学习领域。
虚拟筛选的应用介绍。
新兴虚拟筛选结合深度学习的研究热点(DTA预测)。
DeepDTA、WideDTA、GraphDTA等预测方法的出现。
GNN(图神经网络)的背景:图神经网络(GNN)在社交网络、知识图、推荐系统甚至生命科学等各个领域得到了越来越广泛的应用。GNN在对图节点之间依赖关系进行建模的强大功能,使得与图分析相关的研究领域取得了突破。在DTA领域,GraphDTA采用CNN提取蛋白质序列表示,并在分子图上实现GNN模型,提高了DTA的预测性能。
GraphDTA:
而在GraphDTA中,利用CNN通过序列获取蛋白质特征,并没有为每个蛋白质构建一个图。蛋白质含有大量的原子,如果以原子为节点构造蛋白质的图形,其结构会非常庞大,训练成本非常高。如果以残基为节点构造蛋白质的图,则所构造的图只是一个由肽键连接的长链,不能作为一个图进行计算。因此,通过蛋白质序列构建蛋白质图是一个有待解决的问题。
引入蛋白质接触图contact map
因此适合构造以残基为节点的图。然而,残基之间的连接只是一个长链,没有任何空间信息。因此,本文介绍了接触图(contact map)。接触图是蛋白质结构的一种表示,它是三维(三维)蛋白质结构的二维(二维)表示,它被用作蛋白质结构预测的输出。更重要的是,输出接触图,通常是一个矩阵,与gnn中的邻接矩阵完全一致,这提供了一种将两个数据源组合在一起的有效方法。
Pconsc4------一个接触图预测方法(工具)
Pconsc4是一种快速、简单、高效的接触图预测方法,其性能与目前最先进的方法相一致。因此,本文介绍Pconc4来构建蛋白质接触图和蛋白质图。
DGraphDTA
文章提出DGraphDTA,利用contact map,将gnn同时应用于蛋白质和分子图上,以提高性能,并在基准数据集中获得了良好的预测结果。
模型概念架构:
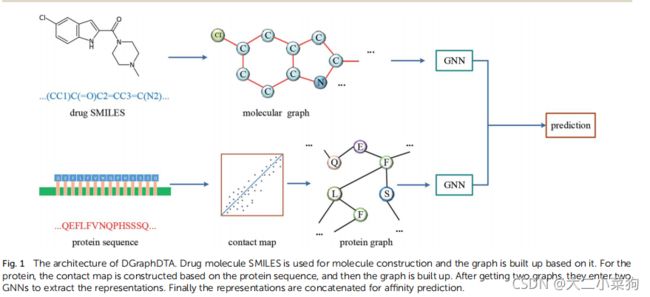
数据集
采用DeepDTA提出的基准数据集进行性能评估。基准包括Davis和KIBA数据集。
(由于对下面这些解释不是很明白所以放了原文)
Davis
Davis数据集包含了来自激酶蛋白家族和相关抑制剂的特定条目以及它们各自的解离常数Kd值。
The Davis dataset contains selected entries from the kinase protein family and the relevant inhibitors with their respective dissociation constant Kd values
KIBA
KIBA数据集包含来自不同来源的联合激酶抑制剂生物活性,如Ki、Kd和IC50,生物活性使用KIBA评分进行处理,KIBA评分用于训练和预测。
The KIBA dataset contains combined kinase inhibitor bioactivities from different sources such as Ki, Kd and IC50 and the bioactivities are processed using KIBA score which is used for training and prediction.
数据集文本截图
数据集信息表

解离常数计算公式

在基准测试中,每个数据集被分为六个部分,一个用于测试,剩下五个用于交叉训练和验证。
药物图构建与节点特征选择
在数据集中,一个亲和条目包含一个分子-蛋白质对。药物分子是用微笑来描述的。在该方法中,以原子为节点,键为节点,作为边缘,构建分子图。为了保证在图卷积过程中能够充分考虑节点的特征,还在图构建中添加了自环,以提高药物分子的特征性能。该分子的图结构如图所示。


蛋白质图构建与节点特征选择
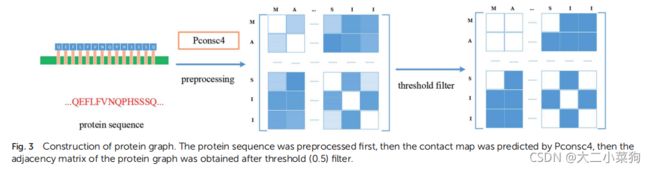
由于蛋白质图和特征的生成取决于蛋白质序列比对结果,因此引入了预处理,包括序列比对、序列筛选等步骤,如下图所示。

蛋白质结构预测:
蛋白质结构预测的目的是根据蛋白质序列来分析和构建蛋白质的三维结构。蛋白质的结构信息包含了不同残基对的连接角度和距离。接触图是结构预测方法的一种输出,通常是一种矩阵。
接触图的形式
假设蛋白质序列的长度为L,那么预测的接触图M是一个有L行和L列的矩阵,其中M的每个元素mij表示对应的残基对(残基i和残基j)是否接触。一般来说,如果两个残基的Cb原子(甘氨酸的原子)之间的欧氏距离小于光谱阈值,则认为两个残基是接触的。
阈值:由于蛋白质结构预测输出的结果是残基之间接触的概率,概率大于阈值才算接触
Pconc4是一种快速、简单、高效的接触图预测方法,其性能与目前最先进的方法相一致。因此,本文介绍Pconc4来构建蛋白质接触图和蛋白质图。
特征选取解释
在得到蛋白质的邻接矩阵时,需要提取节点特征进行进一步处理。由于该图是以残基为节点构造的,所以应该围绕残基选择该特征,因此由于不同的R组,该特征显示出不同的属性。这些性质包括极性、电阳离子、芳香性等。此外,PSSM是蛋白质组学中蛋白质的常见代表。
进化特征
PSSM:
在PSSM中,每个残基位置可以根据序列比对结果进行评分,用于表示残基节点的特征。实现了其简单的PSSM计算。首先,通过计算每个位置的出现,创建一个基本位置频率矩阵(PFM),如eqn(2)所示:
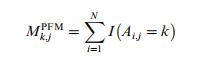
其中A是一个长度为L的蛋白质序列的N个对齐序列集,k属于残基符号集, i = (1, 2, …,N), j = (1,2,…,L),I(x)是条件满足时的指示函数,否则为0。
PFM:
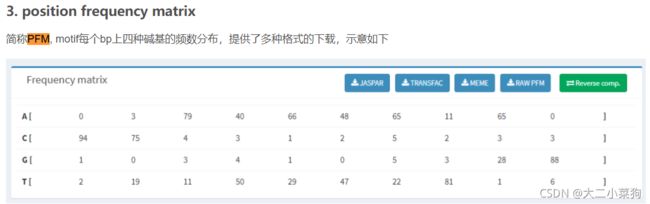
然后利用eqn(3)可以得到一个位置概率矩阵(PPM)54:

其中,p是添加的伪计数(pseudocount),以避免值为0的矩阵元素,并将其设置为0.8。然后,使用PPM作为PSSM来表示剩余节点的一部分特征。
在运行Pconsc4程序进行蛋白质预测与计算PSSM时,它们的input是蛋白质序列比对(protein alignment)的结果。因此,在预处理阶段,基准数据集中所有蛋白质的比对需要在最开始完成。为了提高计算速度,采用HHblits来实现蛋白质序列比对。之后,通过HHfilter和CCMPred脚本在比对结果上获取PSICOV格式的比对结果。
hhsuite (https://github.com/soedinglab/hh-suite)
ccmpred (https://github.com/soedinglab/CCMpred)
综上所述,本文利用54位特征来描述剩余节点。这些特性的细节如表3所示。然后,节点特征的形状为(L、54)。并通过GNN对邻接矩阵和节点特征进行处理,得到相应蛋白质的向量表示。

模型体系结构:
GNN已经衍生出了许多强大的变体,如GCN和GAT。这些模型对于图的特征提取非常有效。对于GCN,每一层都将通过eqn(4)进行卷积操作:

gnnhttps://www.bilibili.com/video/BV1Tf4y1i7Go
这个up还讲了GCN(图卷积神经网络)与GAT(图注意力神经网络)
DGraphDTA总体架构:
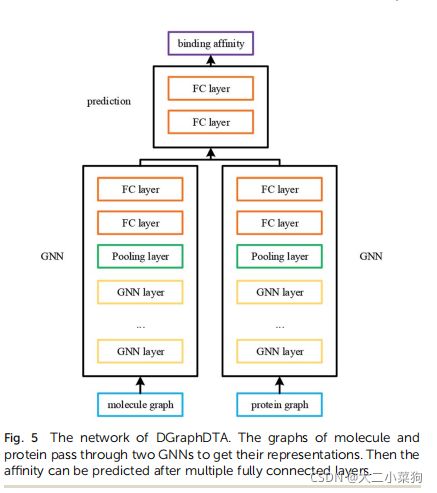
在本文的实验中,我们发现利用三层卷积网络提取小分子和蛋白质的特征是最有效的。
对模型性能优化的解释:
药物分子
对于小药物分子来说,组成分子的原子通过共价键连接,不同的原子和结构最终将表现为不同的分子性质,并通过连接与外部世界相互作用。因此,利用图卷积,充分考虑了这些不同原子之间的关系,从而有效地提取了分子的表示。
蛋白质
对于蛋白质图,使用另一个GNN来提取表示。在蛋白质结构中有很多的空间信息,这对蛋白质和分子的结合亲和力具有重要意义。通过结构预测方法得到的蛋白质接触图可以提取每个残基的信息,主要是基于残基对的相对位置和相互作用。这些残基对的相互作用可以通过GNN获得的载体来充分描述蛋白质的空间结构。在计算机辅助药物设计中,仅通过序列获得蛋白质的表示是一项困难的任务。利用GNN,DGraphDTA可以将蛋白质序列映射到具有丰富特征的表示上,为蛋白质的特征提取提供了一种有效的方法。该方法利用Pconsc4在只了解序列的前提下构建蛋白质的拓扑结构,发现蛋白质整个结构的隐藏信息,有助于亲和预测。
其他因素
此外,影响网络结构性能的因素也很多,如网络层数、GNN模型的选择和Dropout的概率。由于训练过程需要大量的时间,一些超参数是由人类的经验选择的。对于其他重要的超参数,在实验部分进行了比较和确定。
对于每个分子和蛋白质的图,每个节点的特征维数是固定的,但每个图的节点数量不是固定的,这取决于原子或残基的数量。因此,GNN输出矩阵的大小随着节点数量的增加而变化,在两个GNN后添加全局池,以确保对于不同节点数的蛋白质和分子可以输出相同的表示大小。假设最后一个GNN层输出具有形状的蛋白质表示,则全局池可以计算为:
