Chapter1 : Application of Artificial intelligence in Drug Design: Opportunity and Challenges
reading notes of《Artificial Intelligence in Drug Design》
文章目录
- 1.Introduction : What Challenges Does Drug Design Face
- 2. Application of Artificial Intelligence in Drug Design
-
- 2.1.Virtual Screening
-
- 2.1.1.Introduction
- 2.1.2.Dataset Bias in Machine Learning-Based Virtual Screening
- 2.1.3.Receptor Structure-Based Virtual Screening (Docking)
- 2.1.4.Ligand-Based Methods (QSAR)
- 2.1.5.Hybrid VS (Incomprehensible)
- 2.1.6.Opportunities
- 2.1.7.Conclusions
- 2.2.Computer-Aided Synthesis Planning (CASP)
-
- 2.2.1.Introduction
- 2.2.2.Rule-based CASP
- 2.2.3.Reaction Data
- 2.2.4.ML-based CASP
- 2.2.5.Predicting New Chemical Reactions
- 2.2.6.Integration with Robotics
- 2.2.7.Opportunities
- 2.2.8.Conclusion
- 2.3.De Novo Molecule Generation
-
- 2.3.1.Introduction
- 2.3.2.Requirements of De Novo Molecule Generation
- 2.3.3.Requirements of Goal-Directed De Novo Molecule Generation
- 2.3.4.Performance Evaluation of Goal-Directed De Novo Molecule Generation
- 2.3.5.Opportunity
- 2.3.6.Conclusion
- 3.Challenges for Decision Making with Artificial Intelligence in Drug Design
-
- 3.1.Prediction Confidence
- 3.2.Interpretability
- 3.3.Appropriate Validation
- 4.Conclusions
1.Introduction : What Challenges Does Drug Design Face
- R&D cost increased dramatically.
 - Hits with promising potency and ADMET properties are chosen as “lead compounds” —— these then need to be optimized for potency and selectivity while maintaining an appropriate ADMET profile. This process is often ineffective at finding molecules with the right pharmacodynamic and pharmacokinetic properties in patients.- Datasets problem:
- Hits with promising potency and ADMET properties are chosen as “lead compounds” —— these then need to be optimized for potency and selectivity while maintaining an appropriate ADMET profile. This process is often ineffective at finding molecules with the right pharmacodynamic and pharmacokinetic properties in patients.- Datasets problem:
- Many datasets are disproportionately focused on a small range of well-studied endpoints.
- Datasets describing in vitro activities of compounds is larger than those describing their in vivo effects.
- Consistent annotation of data is challenging.
- There is little hope of conducting simulations of complex physiological systems, necessitating the use of empirical models.
2. Application of Artificial Intelligence in Drug Design
2.1.Virtual Screening
2.1.1.Introduction
- HTS(high-throughput screening) screens large chemical libraries against project relevant activity assays.
- VS(virtual screening) attempts to address the shortcomings of HTS by screening compounds in silico rather than in vitro.
- VS can be categorized into two types: ligand-based VS and structure-based VS.
- ligand-based VS uses a set of compounds that are known to be active and tries to identify other molecules. This needs a predictive model to prioritize active compounds.
- structure-based VS evaluates a ligand based on the complementarity of 3D structures with the target’s binding pockets. This needs 3D information which is hard to obtain.
2.1.2.Dataset Bias in Machine Learning-Based Virtual Screening
- Molecular datasets used in VS incorporate bias due to several reasons.
- Their size is comparably small relative to the space of potential molecules.
- The nature of drug development pipelines leads to bias : synthesis efforts typically focus around known successful molecules.
- Novel molecules are often proposed in series that grow during hit selection and lead optimization. As a result, explored regions of chemical space are formed of clusters rather than uniform samples.
- The practitioners should be careful about how they split their dataset for training and testing. (Butina-Taylor)
- Another caveat that practitioners should be aware of is the lack of universally accepted chemical datasets on which to evaluate models for VS. (DUD-E, MUV, AVE)
- Bias is not always undesirable.
2.1.3.Receptor Structure-Based Virtual Screening (Docking)
- Most ML approaches to structure-based VS have focused on improving the scoring function. Gnina and AtomNet have develop their own standalone scoring functions. Other ML model (such as △VinaRF) for scoring attempt to improve rather than replace.
- One way to improve scoring functions is to tailor them to the problem of interest. ML model is more suitable than traditional docking algorithms.
- An important limitation of current ML model with respect to traditional docking algorithms is that they generally lack the capabilities to produce docking poses, so they rely on external software to obtain them. This means ML model performance is capped by the performance of external pose generation.
2.1.4.Ligand-Based Methods (QSAR)
-
What characterizes ligand-based VS is that it does not utilize any information about the receptor. Therefore, it can be applied to general problems.
-
QSAR and ML model is similar: both are supervised methods that identify pattern in molecular data to learn a target signal.
-
In addition to fitting complicated target functions, ML model can allows a greater breadth of molecular representations, some of them is very abstract but important such as molecule-induced transcriptomic signatures or cell painting imaging profiles.
-
In addition to using new biological data types, another advantage of some ML model is that they can learn their own representation from a dataset. VAE’s latent space is usually smaller in size than the input, so the model is forced to find a compressed representation of the input.
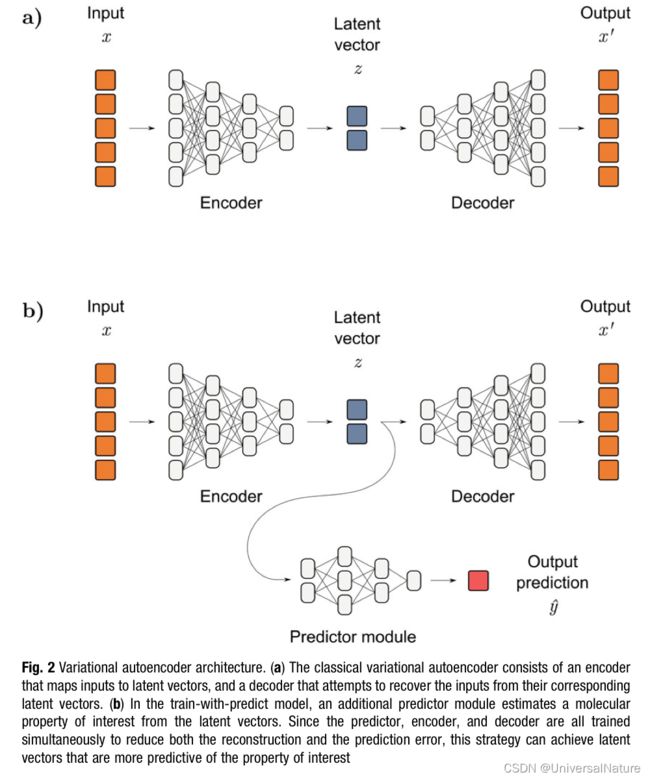
-
Some ML models accepted flexible training regimes which presents another opportunity for improvement of ligand-based. There are several methods based on the idea of shared knowledge, such as transfer learning, multitask learning, self-supervised learning.

-
Flexibility brings practical difficulties, too. The high degree of choice makes it difficult to decide what model is most promising for a specific problem. The part of challenge is that it is difficult to diagnose why a ML method is successful.
2.1.5.Hybrid VS (Incomprehensible)
- Hybrid VS refer to statistic al models for bioactivity prediction that incorporate information about the receptor, but in more abstract form than a geometric description of the binding pocket. It is often considered an expansion of ligand-based VS and QSAR.
- The most common type of Hybrid VS are proteochemometirc models (PCM).
- Many of the opportunities and limitations discussed in Ligand-Based VS is applicable here.
- PCM is the flexibility and customizability of input representations. DeppDTA and DGraphDTA could produce target representations.
- PCM is building models that scale to the big-data regime, such as collaborative filtering.
- ML is helpful for PCM in uncertainty quantification. A work that applied a Gaussian process to PCM found that Bayesian model achieved calibrated variance estimates for each prediction.
- ML could be used to design novel hybrid workflows different from PCM, such as DeepDocking.
2.1.6.Opportunities
- An promising area of research is the aggregation of data from different sources to facilitate learning in the low-data regime, such as transfer learning and multitask learning.
- Another area may be productive for VS is the utilization of unlabeled data.
- Another avenue that could bring substantial improvement to VS is learning from new types of data.
2.1.7.Conclusions
- ML methods can outperform traditional VS in a range of applications, but special care should be taken in benchmark evaluations to ensure the results obtained are generalizable.
2.2.Computer-Aided Synthesis Planning (CASP)
2.2.1.Introduction
- On average, the chemical synthesis of a design-make-test-analyze (DMTA) can take 8-12 weeks, most significant contributing to overall DMTA cycle time. Furthermore, if a molecule is successfully progressed to the clinical stages of drug discovery, synthesis must be scaled up and cost, time of synthesis becomes pivotal.
- There are some key principles in retrosynthesis:
- whether reactants will form the desired product
- to what extent the product will be formed
- finding the optimal reagents and condition used to carry out the reaction
- whether the selected retrosynthesis disconnection is the best strategy
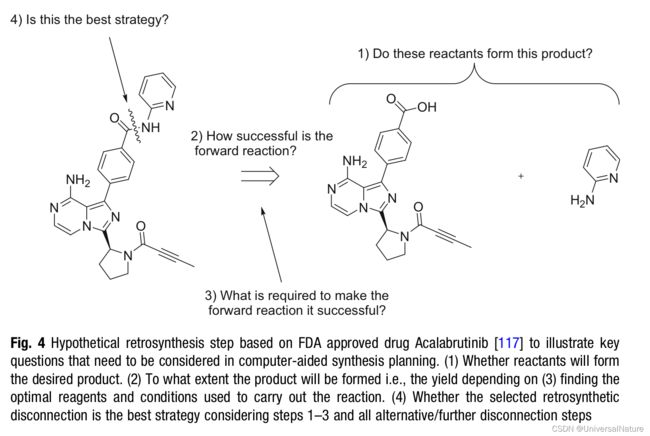
- AI-based CASP could be categorized into older and more established rule-based methods and ML-based methods.
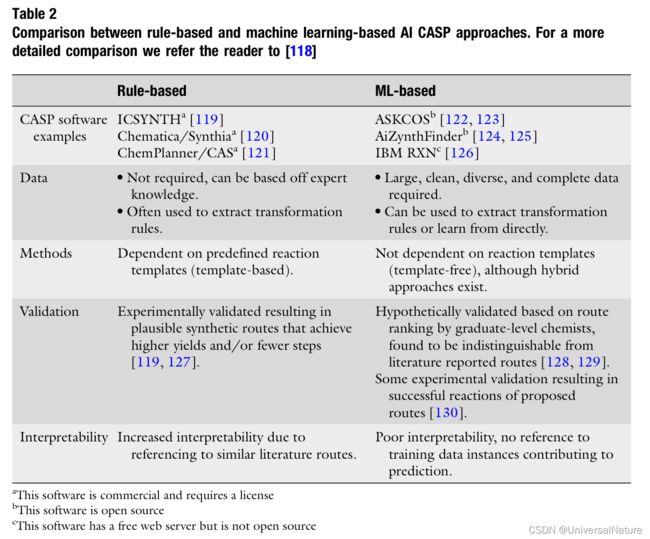
2.2.2.Rule-based CASP
-
Early rule-based CASP programs used hand-coded transformation rules describing reaction centers and functional groups that may affect reactivity. Succeeding rule-based CASP programs instead automatically extract transformation rules from reaction databases.
-
Rule-based CASP programs have undergone relatively thorough prospective validation evidencing successful application.
2.2.3.Reaction Data
- The integration of ML-based models into CASP is inherently restricted by reaction data availability and quality.

- Electronic laboratory notebooks (ELN) —— reaction data discussed thus far is likely to exhibit a bias towards successful reactions, however, negative data is crucial to building balanced datasets for ML algorithms.
- The sparsity and inherent bias towards certain reaction types provides an additional challenge for use with ML. This indicates a clear bias towards reproducing what has been done compared to exploring new reaction types and target molecules.
2.2.4.ML-based CASP
-
Forward reaction prediction——ML models need a appropriate output representations that translate to product molecular structure. Coley et al. tackled these problems by firstly modeling this task as predicting the edits made to reaction center atom and secondly augmented chemically plausible negative data. Some other problems were tackled by Coley et al. by using a Weisfeiler-Lehman Network and this model performed on par with human chemists. ML algorithms was inspired by natural language processing that model translation problems circumvent the issue of requiring negative data or reaction templates. The approach used by Schwaller et al. outperforms human chemists on a benchmark set of 80 reactions.
-
Yield prediction——It is much more challenging to predicting how successful the reaction will be in the context of reagents and conditions. Schwaller et al. extend NLP type ML models to predict reaction yields and performance was achievable for clean, curated datasets of specific reaction types, but in worse datasets performance drop significantly.
-
Condition prediction——Some work (e.g., Marcou et al. and Coley et al.) emphasize the difficulty of predicting reaction conditions, regarding not only incomplete data but also the modeling of such flexible parameters, and even difficulty in evaluation of such models.
-
Retrosynthetic strategy——This problem is searching vast potential retrosynthetic possibilities to find suitable synthetic routes. Schwaller et al. proposed new metrics called round-trip accuracy and Segler et al. tackled this problem by using a Monte Carlo tree search (a RL approach) in combination with neural network functions.
2.2.5.Predicting New Chemical Reactions
- Segler et al. presented that inventing new reactions was possible when modeling reaction data as half reactions via heuristic link prediction on molecule-reaction graphs. Other preliminary work suggests that proposing novel routes may be possible via generative models when reactions are considered only as their reaction centers.
2.2.6.Integration with Robotics
- Progress towards automated synthesis execution via robotics was recently shown by Coley et al., who integrated the ASKCOS CASP platform with robotics platform to conduct flow chemistry.
- More progress is required to bridge the gap between CASP and robotics, mostly to include practical aspects of automated synthesis such as risks associated with chemicals, or reaction product identification and purification from a reaction mixture.
2.2.7.Opportunities
- The following significant opportunities for the use of AI in the field of synthesis prediction seem to emerge currently:
- new technologies such as “digital glassware” may improve both the quality and quantity of reaction data
- the development of CASP tools geared towards further methods of synthesis,such as biocatalysis
- sufficient and universally accepted benchmarks for the prediction of retro and forward synthesis are lacking
2.2.8.Conclusion
- The progression of new ML-based AI methodologies in CASP is significantly restricted by the access to and quality of reaction data, which is often sparse, noisy, partially incorrect, and incredibly bias. Realistically speaking, CASP is——as is computational methods in other areas—unlikely to replace experimental chemists, although it appears that quite possibly “chemists who use AI will replace those that do not”.
2.3.De Novo Molecule Generation
2.3.1.Introduction
-
The most challenging aspects of of de novo molecule generation, is that in many cases the endpoint is not explicitly known. Conceptually, this question is usually addressed by an iterative design process known as the design-make-test-analyze (DMTA) cycle.

-
As described by Schneider et al., earlier models had to address the three main objective: how to build a chemical structure, how to evaluate molecule quality and how to optimize chemical space efficiently. One example is the growing of structures by fragments (generation) conditional upon a receptor binding pocket, its steric constraints and hydrogen bonding sites (evaluate) and using a depth or breadth-first algorithm to explore possibilities (searching), as in Skeletons.

- The generation model could be categories into two types: rule-based models where chemical structure building instructions are hard coded and distribution-based models where chemical structure generation is learned from data.
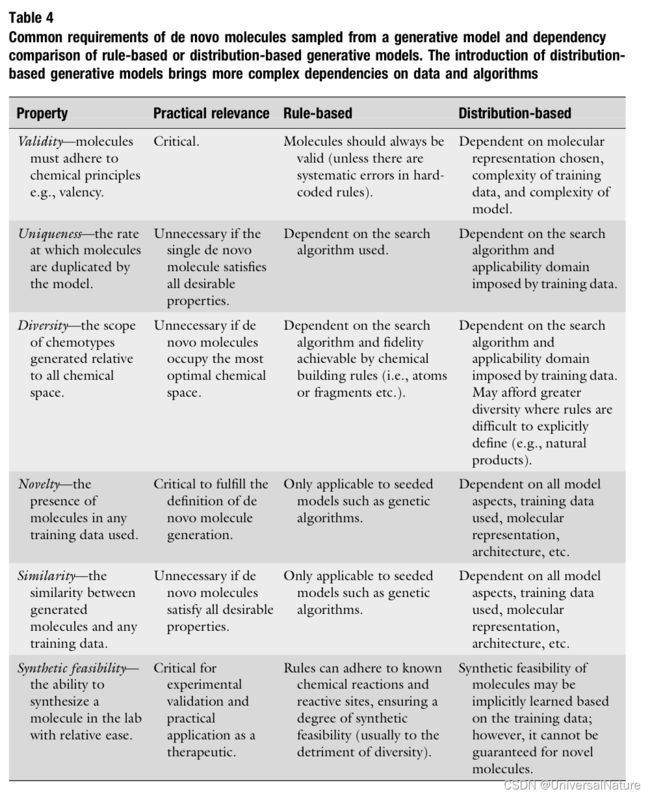
2.3.2.Requirements of De Novo Molecule Generation
- Universal to all generation models, they must perform well at the basic task of generation molecules which Table4 has summarized. However, in practice, these properties are challenging to measure.
- Better descriptive metrics that can better capture more meaningful properties of molecules and distinguish between well performing complex models and naive models would significantly benefit the field.
2.3.3.Requirements of Goal-Directed De Novo Molecule Generation
- Suitable drug candidates must possess necessary other properties (examples shown in Table5).


-
A common theme in scoring function pitfalls, that is also relevant outside their use in generative models, is the large disconnect between property endpoint and scoring function proxy.
-
Behavior of generative models when optimizing towards scoring function can further exacerbate scoring function limitations. At some point during training, the model is likely to evaluate molecules outside its domain of applicability resulting in aberrant predictions, hence evaluating model confidence is so important.
-
The ability to optimize all desired properties simultaneously in a multiparameter/multi-objective optimization (MPO/MOO) is one of difficulty and often neglected in publications.
2.3.4.Performance Evaluation of Goal-Directed De Novo Molecule Generation
-
Figure7 demonstrate the difficulties of generation model performance measurement.
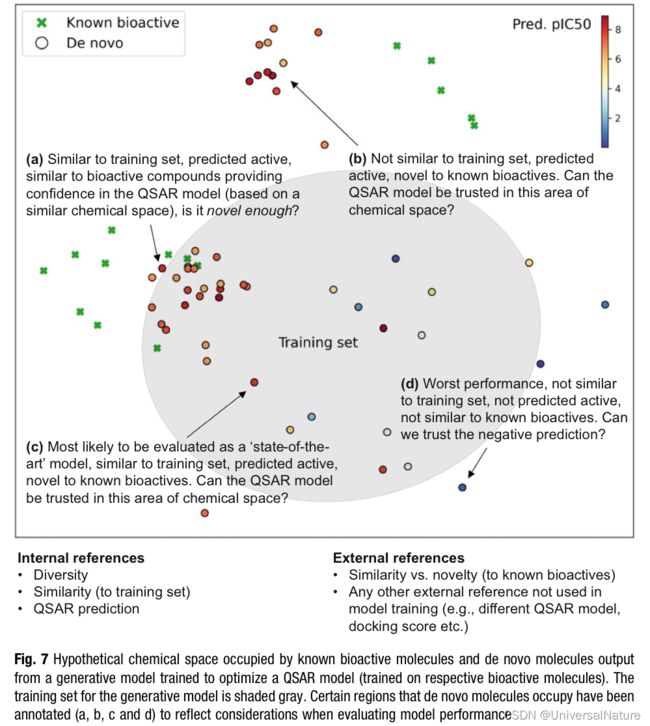
-
Furthermore, molecules could likely be found using traditional drug design approaches, raising further concern as to the real world of generative models over traditional methods.
-
Model evaluation should focus more effort in the context of prospective application, so that evaluation is more interpretable when considering integration of de novo design with generative models into real-world projects.
-
It’s difficult enough to robustly measure individual properties such as molecular diversity. However, improvements are being made in establishing more interpretable metrics, such as Zhang et al. 's GBD-13.
-
It can also be unclear to what extent model evaluation is due to expert intervention, such as Zhavoronkov et al. 's model named GENTRL. This aspect is often overlooked.
-
Understanding how generative models compare to other non-AI methods is yet to be determined.
2.3.5.Opportunity
- The first obvious step beyond most current generative models is the leap from 2D topology to 3D molecular structure.
- Generative models could also benefit from increased research in more robust and more efficient scoring functions.
- New generative models that are highly efficient in optimizing external scoring functions may provide new opportunities to understand scoring function behavior itself.
- It would be also be beneficial to better understand the co-dependent behavior of generative models and optimized scoring function.
- The field could certainly benefit from stronger model evaluation and comparison.
2.3.6.Conclusion
-
The premise of generative models is accelerated and higher quality drug design in silico. The former has some evidence, while the latter is still an open research goal with greater potential impact.
-
It is also worth noting that potentially improved efficiencies because of the use of generative models may result in less experimental screening, which could negatively impact data collection—data availability (for optimization) being one of the current limiting factors to current generative models.
3.Challenges for Decision Making with Artificial Intelligence in Drug Design
- Many AI systems, especially those which rely on deep neural networks, despite their high predictive performance, are liable to catastrophic failures——this limits their use in decision making when the consequences of wrong decisions are severe.
- Expert oversight can be used to check the model decision process, increasing reliability.

3.1.Prediction Confidence
-
The predictions of ML models are subject to multiple sources of heterogeneous errors.
- Supervised ML methods generally assume that feature inputs will be drawn from a distribution identical to the training data.
- Many target variables of interest in drug design are inherently uncertain because of biological and experimental variability.
- These problems become more pronounced when dataset from different labs or different assays are combined.
-
Predictive uncertainty can be split theoretically into two components: aleatoric uncertainty and epistemic uncertainty.
-
In order to identify prediction with high epistemic uncertainty, model application domains can be defined.
-
In order to account for variations in uncertainty across the training data, the boundaries of the domain can be refined by considering the local performance of the model.
-
Estimated the total prediction uncertainty of a model is important. Given a model, there exist frequentist methods for uncertainty prediction which transform the model’s predictions to give predictive uncertainty, using a held-out validation set, such as conformal regression and Venn-ABERS approach.
-
As an alternative to these frequentist methods for uncertainty estimation, Bayesian models can be used.
3.2.Interpretability
-
Manual checking of prediction for concordance with external data and literature is needed. As a result, it is necessary for a user to understand how and why a model has made a particular prediction.
-
The predictions of an interpretable model need to be reducible to a small number of key parameters.
-
As an alternative to construction of interpretable models, external explanation methods which associate selected inputs and outputs of complex models to construct a simpler “metamodel” have been proposed.
-
One way of interpreting an ML model in drug design is to assess which features of an input example have most strongly affected a model’s decision; this is known as feature attribution. The ease with which this can be done depends on the model used.
- linear models is trivial to extract this information
- deep neural network can be studied using gradient-based methods, such as Likelihood Relevance Propagation
- Attention-based neural network are more interpretable.
-
Another way in which AI systems can justify their decisions is to link input examples to relevant training examples. Evaluating similarity over the representations learned by neural networks can be used to identify molecules that are processed similarly by the neural network; this is a promising way to identify potential analogues. To justify inference of the properties of an unknown chemical, structurally similar chemicals with known properties must be presented, and the relationship between structure and properties for these molecules must be described
3.3.Appropriate Validation
- The need for uncertainty quantification and interpretability reflects practical considerations for model use; however, they have been relatively neglected in research into ML in drug design.
4.Conclusions
-
The potential of AI in drug design is slowly approaching reality, but much work remains to be done
- it is essential that models prioritize interpretability and applicability over raw predictive performance
- many AI methods are evaluated on irrelevant tasks or with inappropriate metrics
- user testing of models by medicinal chemists may help to evaluate models for practical usefulness
-
There are several applications of AI to drug design which are yet to achieve their full potential
- the multiparameter optimization of molecules by generative models
- integration of AI with structure-based methods such as protein structure prediction, docking simulations and free energy calculations to improve accuracy and computational efficiency