如何在Gromacs中添加自定义溶剂
如何在Gromacs中添加自定义溶剂
- 在Gromacs中添加自定义溶剂
-
- 准备自定义溶质的拓扑文件
- 在Gromacs中添加自定义立场
- 使用Gromacs生成水层的top文件
- 将水溶于CH4
- 修改top文件中molecules数量
- 能量最小化
- NVT预平衡
- NPT预平衡
- MD成品模拟
在Gromacs中添加自定义溶剂
Gromacs中的默认溶剂一般只有水溶液,但有时候需要在Gromacs中将蛋白融入非水溶剂,例如将C的化合物融入有机溶剂,下面将介绍如何在Gromacs中自定义溶剂,并制作自己的仿真盒子进行MD模拟。具体实例将水融入CH4中,随后对水进行MD模拟。
准备自定义溶质的拓扑文件
这里我用Gaussview画了一个碳原子,另存为PDB文件,Gaussview会自动补充氢原子
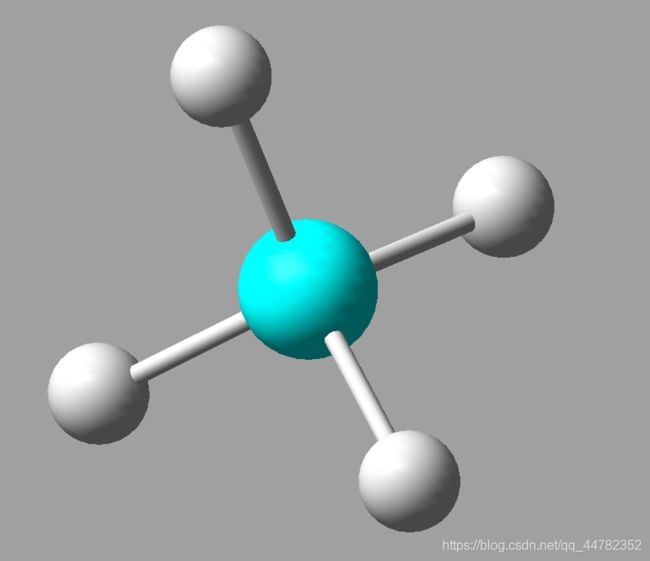
随后我们需要在准备这个结构的.itp文件和.gro文件,这里我用的是LigParGen(http://zarbi.chem.yale.edu/ligpargen/)自动生成这个原子的两个文件,文件放在后面了,想重复的可以下载。
在Gromacs中添加自定义立场
准备好分子的拓扑文件后,还需要将文件夹放在Gromacs的力场文件中,这里我使用全原子立场OPLS-AA,所以先找到Gromacs安装文件中的oplsaa.ff文件夹,一般是在/share/top/中,在这里复制自己需要的力场文件夹到工作文件夹,这里有的教程写的是可以在原文件夹中添加自定义文件,再在命令中加上-name选择这个文件,我的Gromacs版本是2018.1,经过测试这种方法在这个版本中并不可行。
然后在OPLS-AA夹和当前文件夹 中,将Carbon.itp和Carbon.gro文件复制进去。
使用Gromacs生成水层的top文件
众所周知,Gromacs有三大输入文件.gro .itp(.top) .mdp
所以需要通过pdb文件得到.gro与.top文件
在终端依次中输入
gmx editconf -f water.pdb -o water.gro -c -d 1.0 -bt cubic
gmx x2top -f water.gro -o water.top -ff select -nopbc -kb 400000 -kt 600 -kp 150

出现让选择力场的提示,选择1,这是我们刚才自己构建的力场。
每次成功Gromacs都会reminds you。
将水溶于CH4
这里的溶于指在仿真盒子中以水分子为中心,周围均匀填充CH4,注意如何你的蛋白中间的空隙如果足够填充一个溶剂分子的话,溶剂分子会进入蛋白的结构。如果不希望这种情况的发生,则需要规定溶剂移动范围。
在linux终端中输入
gmx solvate -cp water.gro -cs Carbon.gro -p water.top -o conf.gro

得到这样的结果说明溶解成功。
修改top文件中molecules数量
由于Gromacs自带的代码中只能自动添加水溶剂的molecules数量,所以我们需要在top文件中自己添加溶剂的数量。
首先我们先尝试着输入一条能量最小化的命令
gmx grompp -f minim.mdp -c conf.gro -p water.top -o em.tpr
由于这时候gro文件与top文件中的分子数量不匹配,Gromacs会报错
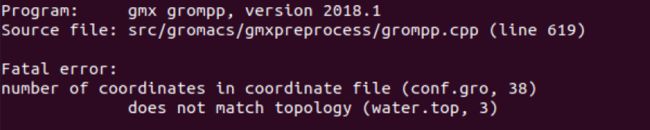
这时候我们就要计算一下要添加的分子数量,由pdb文件可知,一个CH4分子有5个原子,而从报错信息中可以看到,现在top文件中只有3个原子,而gro文件中有38个原子所以需要添加的原子为
(38-3)/5 =7
首先用文本格式打开carbon.itp文件

在Name这里找到itp文件中的moleculetype的名字,这里建议改成和实际原子对于的。
然后用文本格式打开water.top文件,在文本的最后添加这个分子名字和上面计算的分子个数。

这里我直接用默认名称了,不建议这样。
记得还要在立场文件的定义下面再加一行自己定义分子的itp文件
#include "./oplsaa.ff/carbon.itp"
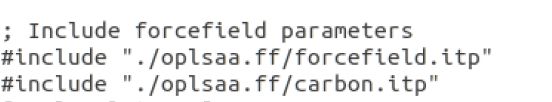
然后就可以正常进行能量最小化了。
能量最小化
依次输入
gmx grompp -f minim.mdp -c conf.gro -p water.top -o em.tpr
gmx mdrun -v -deffnm em
NVT预平衡
依次输入
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p water.top -o nvt.tpr
gmx mdrun -v -deffnm nvt
NPT预平衡
依次输入
gmx grompp -f npt.mdp -c nvt.gro -r nvt.gro -p water.top -o npt.tpr
gmx mdrun -v -deffnm npt
MD成品模拟
依次输入
gmx grompp -f md.mdp -c npt.gro -r npt.gro -p water.top -o md.tpr
gmx mdrun -v -deffnm md
成品模拟和就可以计算需要计算的参数了,MD模拟的参数mdp文件也放在下面了需要的可以自行下载。
注意由于版本不同可能会导致一些bug,如果有问题可以在下面回复,看到我会一一回复,例如有时候用editconf自动生成的gro文件的尺寸不合适,这时还需要人工修改.gro文件中仿真盒子的尺寸。
所有的资源在 这里