Paper再现:MD+AI自动编码机探测蛋白变构(二):MD数据处理及特征化
2.1 数据的预处理
先来讲讲项目的结构吧。
整个项目的文件路径如下图。在dataset文件夹保存了模型训练所需要的全部数据,包括original_dataset和dataset两个文件夹。original_dataset保存的是MD生成的PDB结构,是datset split.ipynb进行数据清洗分割以后的结果,分为两个holo和opo两个文件夹,其中opo就是文献中提及的Unbound的MD的结构快照,共计1000个,文件名分别为0.pdb,1.pdb, 2.pdb,3.pdb …. 999.pdb;holo就是文献中提及的bound的MD的快照,共计1000个,0.pdb,1.pdb, 2.pdb,3.pdb …. 999.pdb。因此,我们复现模型的时候,每一个残基对的输入向量的长度是1000。同时,我们并没有将opo、holo分别做了两次,所以,我们只有opo-holo一个模型。dataset文件夹保存的是Protein PDB Preprocess.ipynb进行数据转化和预处理之后,保存的矩阵数据,可以直接输入到自编码模型中。
由于数据的清洗和分割是依项目而异的情况,因此,这里就不介绍datset split.ipynb文件的处理过程。接下来就是数据的预处理。

按照文章的要求,模型输入的是两两残基对之间的距离,所以要先计算两两残基之间的距离,使用残疾序号作为残基的标识。下面分别将holo和opo的PDB分析,清理出我们需要使用的数据。
导入相关的模块:
import os
import numpy as np
import pandas as pd
from scipy.spatial.distance import cdist
2.1.1 读取PDB文件信息
首先需要介绍一下PDB文件。PDB文件相当于一个txt文件,PDB文件里面的每个记录都有着严格的格式. 每个记录中的字段, 如标识, 原子名称, 原子序号, 残基名称, 残基序号等, 不仅要按照严格的顺序书写, 而且每个字段所占的字符串长度, 及其所处的位置都是严格规定好的。所以,文件中的每一行是对齐的,每一个字符都代表特定的信息,如下图。每一行都代表一个原子,分别记录了记录类型,原子序号,原子名字等信息。
其中,前4个字符代表的是记录的类型,对于残基,其记录类型都是ATOM。
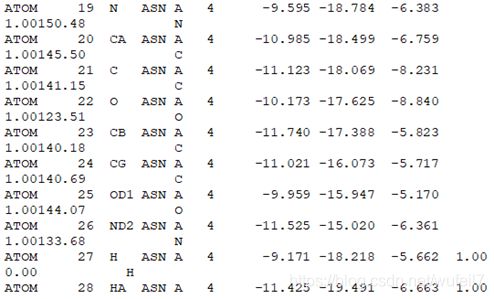
至于每一个字符分别代表什么请见下图。关于PDB更多的内容,请见:https://jerkwin.github.io/2015/06/05/PDB%E6%96%87%E4%BB%B6%E6%A0%BC%E5%BC%8F%E8%AF%B4%E6%98%8E/。

接下来,创建一个读取PDB里面所有信息的函数,将PDB文件保存成我们熟悉的Dataframe格式。get_all_pdb_info函数实现了这么一个功能。将一个PDB文件输入,使用打开文本文件的方法直接打开,然后逐行读取。
def get_all_pdb_info(PDB):
'''
获取PDB文件内的所有残基原子和杂原子的信息
:param PDB: pdb文件名
:return: dataframe格式
'''
with open(PDB, "r") as pdb_file:
pdb_content_list = pdb_file.readlines()
# 抽取数据
columns = ['RecordType', 'AtomNumber', 'Name', 'AltLoc', 'resName', 'chainID',
'ResSeq', 'X', 'Y', 'Z', 'Occupancy', 'TempFactor']
result = []
for line in pdb_content_list:
if line[:6] in ['ATOM ', 'HETATM']:
try:
'''
这里的try和except的目的是为了解决在MD过程中会产生大量的特殊编码的水分子,int(line[22:26])会报错
'''
line_i = [line[:4], int(line[6:11]), line[12:16].strip(), line[16],
line[17:20], line[21], int(line[22:26]), float(line[30:38]),
float(line[38:46]), float(line[46:54]), float(line[54:60]),
float(line[60:66])]
result.append(line_i)
except:
pass
# print('Below pdb line is not standar pdb format, pass ... ... \n',line)
result = pd.DataFrame(result, columns=columns)
result['index'] = [i+'-'+str(j) for i, j in zip(result['chainID'], result['ResSeq'])]
result = result.drop_duplicates(keep='last')
return result
由于在模型中,仅仅考虑了残基对的距离的扰动,没有考虑水分子,配位分子中的原子,因此,我们仅仅保留前4个字符是ATOM的行(原子)。同时,根据上图及链接的说明,将信息按列保存到dataframe中。
先输入2个PDB文件试试。
holo = 'dataset/original_dataset/holo/0.pdb'
opo = 'dataset/original_dataset/opo/0.pdb'
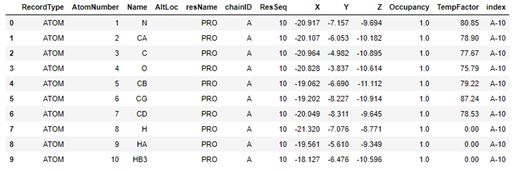
opo_data = get_all_pdb_info(opo)
holo_data = get_all_pdb_info(holo)
print('opo和holo的数据长度为:{}, {}'.format(len(opo_data), len(holo_data)))
opo_data.head(10)
输出为:opo和holo的数据长度为:6987, 7351。
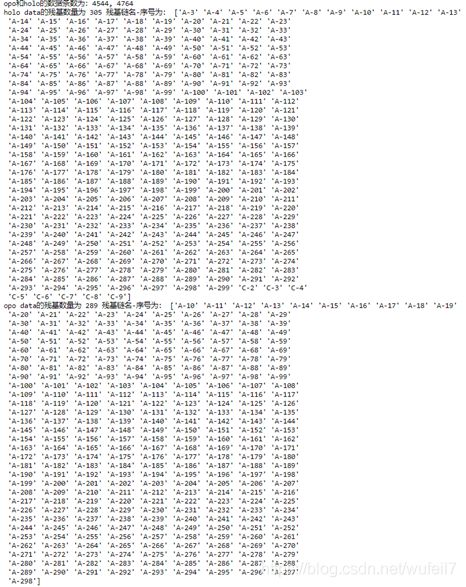
查看各条原子信息对应的残基序号、个数及其链名,也就是对dataframe进行简单的操作:
#残疾个数及其序号
holo_data_ = holo_data.drop_duplicates(subset='index', keep='first')
holo_index = holo_data_['index'].values
print('holo data的残基数量为',len(holo_index),'残基链名-序号为:', holo_index)
opo_data_ = opo_data.drop_duplicates(subset='index', keep='first')
opo_index = opo_data_['index'].values
print('opo data的残基数量为',len(opo_index),'残基链名-序号为:', opo_index)
输出为:
2.1.2 计算残基质量中心
注意一下,按照文章的要求,在计算残基对距离的时候,距离指的是两两残基质量中心的距离。所以在一个PDB文件(快照)里面,要先计算残基的质量中心,然后在计算残基对之间的距离。所以,先写一个计算残基质量中心坐标的函数quantity_center,该函数返回的是质量中心XYZ坐标的list。(当然在这个函数中,处理的方法不是很严谨,因为在蛋白的PDB文件中,很有可能是有多条链的情况的,于是残基的序号是存在重复的,那么使用以下的代码显然是不合适的, 请适当修改哈)。
'''
根据文献,计算质量中心!!!
'''
def quantity_center(dataframe, index):
'''
残基的计算质量中心
dataframe:PDB中残基原子信息的dataframe
idnex:需要计算的残基序号
'''
data_need = dataframe[dataframe['ResSeq']==index]
atom = data_need['Name'].values #原子种类列表
x = data_need['X'].values
y = data_need['Y'].values
z = data_need['Z'].values
X = 0
Y = 0
Z = 0
Q = 0
for k in range(len(data_need)):
if atom[k][0] == 'C':
X = X + 12 * x[k]
Y = Y + 12 * y[k]
Z = Z + 12 * z[k]
Q = Q + 12
elif atom[k][0] == 'O':
X = X + 16 * x[k]
Y = Y + 16 * y[k]
Z = Z + 16 * z[k]
Q = Q + 16
elif atom[k][0] == 'N':
X = X + 14 * x[k]
Y = Y + 14 * y[k]
Z = Z + 14 * z[k]
Q = Q + 14
elif atom[k][0] == 'S':
X = X + 32 * x[k]
Y = Y + 32 * y[k]
Z = Z + 32 * z[k]
Q = Q + 32
elif atom[k][0] == 'H':
X = X + 32 * x[k]
Y = Y + 32 * y[k]
Z = Z + 32 * z[k]
Q = Q + 32
X = round(X/Q, 4) #质量中心X坐标
Y = round(Y/Q, 4) #质量中心Y坐标
Z = round(Z/Q, 4) #质量中心Z坐标
return [X, Y, Z]
来试一下:
#计算之前加载的holo数据,第297号残基的质量中心的X\Y\Z坐标
quantity_center(holo_data, index=297)
#计算之前加载的holo数据,第297号残基的质量中心的X\Y\Z坐标
quantity_center(opo_data, index=297)
输出为:([6.34, 16.2165, 24.853], [10.9458, 18.5178, 16.9098])。
2.1.3 PDB文件质量中心特征化
现在尝试着对一个蛋白快照(PDB文件)进行处理, 获取蛋白每一个残基的中心坐标,写成一个函数pdb_feature吧,它的输入是一个PDB文件,自行调用上述的获取原子信息,提取残基原子(舍弃水分子等中的原子),最后计算出质量中心的字典,其中残基的链名-序号为键,值为质量中心。函数代码如下:
'''
现在尝试着对一个蛋白进行处理, 获取蛋白每一个残基的中心坐标
'''
def pdb_feature(pdb):
#获取残基和杂质原子的信息
data = get_all_pdb_info(pdb)
#仅保留残基中的数据,也就是RecordType为ATOM的数据条目
data = data[data['RecordType']=='ATOM']
#计算每一个残基的质量中心
center_coord = {}
for i, index in zip(data['ResSeq'].values, data['index'].values):
center_coord[index] = quantity_center(data, i)
return center_coord
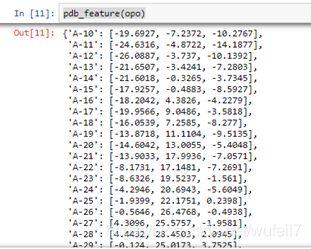
输入一个PDB文件试一下pdb_feature函数:
opo = 'dataset/original_dataset/opo/0.pdb'
pdb_feature(opo)
输出为:
2.1.4 模型输入数据生成
接下来就是直接将所有的PDB都处理好,生成各个残基的质量中心。holo文件夹下的数据处理方式:
#holo文件
holo_dataset = {}
for i in range(1000):
'''
每一个蛋白都保存成holo1_dataset中的一个键值对,
每个残基是值中的字典。{0(snapshot序号):['0':[x,y,x坐标], ...], 1:[...], ... }
'''
holo_dataset[i] = pdb_feature('dataset/original_dataset/holo/'+str(i)+'.pdb')
接下来将holo 1000个PDB文件的质量中心按照文件名的前后组装成一个矩阵,目的是当然是方便接下来的计算。处理成numpy的矩阵,shape为:(1000, len(holo_index), 3),1000代表1000个PDB,len(holo_index)残基的数量,3代表X\Y\Z三个坐标轴。所以,这个矩阵保存了MD过程中,蛋白每一个残基质量中心的位置。当然啦,也保存一份npy的数据格式,方便接续运行。代码如下:
holo_dataset_numpy = np.zeros((1000, len(holo_index), 3)) #残基的坐标
for i in range(holo_dataset_numpy.shape[0]): #snapshot
for j in range(holo_dataset_numpy.shape[1]):#residue index
holo_dataset_numpy[i,j,:] = holo_dataset[i][holo_index[j]]
np.save('dataset/dataset/holo_dataset.npy', holo_dataset_numpy)
print(holo_dataset_numpy.shape)
输出为:(1000, 305, 3)。
接下来终于到了可以计算各个残基对距离的那一步了。残基之间的距离使用scipy里面的cdist函数来计算。所有的快照的残基对距离计算完成以后,得到了距离矩阵。为了形成autoencode的输入结构,距离矩阵进行了轴的交换,形状的重塑,顺路保存了holo_distance_dataset.npy文件。
#计算距残基之间的距离
holo_distance_dataset_numpy = np.zeros((1000, len(holo_index),
len(holo_index))) #残基对距离
for i in range(1000):
snapshot = holo_dataset_numpy[i]
k = cdist(snapshot, snapshot, metric='euclidean')
holo_distance_dataset_numpy[i] = k
#原来是:(snapshot No., 残基序号, 残基序号),
#换成(残基序号,残疾序号,snapshot No)
holo_distance_dataset_numpy = holo_distance_dataset_numpy.swapaxes(0,2)
holo_distance_dataset_numpy = holo_distance_dataset_numpy.reshape(-1, 1000)
np.save('dataset/dataset/holo_distance_dataset.npy',
holo_distance_dataset_numpy)
print(holo_distance_dataset_numpy.shape)
输出为:(93025, 1000)。
npy文件是不能直接知道是哪两个残基对之间的距离,所以尝试将数组保存成CSV的格式(holo_csv.csv文件)。代码的原理需要结合上述的数组转化的流程来理解一下。代码如下:
#保存成csv吧
index = []
for i in holo_index:
for j in holo_index:
index.append(i+' '+j)
holo_csv = pd.DataFrame(holo_distance_dataset_numpy, index=index, columns=range(1000))
holo_csv.to_csv('dataset/dataset/holo_csv.csv')
holo_csv.head(10)
输出结果如下图。第一行代表A3残基与A3残基的质量中心矩阵。
Opo文件的处理是类似的,这里直接上代码:
#opo文件
opo_dataset = {}
for i in range(1000):
'''
每一个蛋白都保存成opo_dataset中的一个键值对,每个残基是值中的字典。{0(snapshot序号):['0':[x,y,x坐标], ...], 1:[...], ... }
'''
opo_dataset[i] = pdb_feature('dataset/original_dataset/opo/'+str(i)+'.pdb')
#处理成numpy的矩阵,shape为:(1000, len(opo_index), 3)
opo_dataset_numpy = np.zeros((1000, len(opo_index), 3)) #残基的坐标
for i in range(opo_dataset_numpy.shape[0]): #snapshot
for j in range(opo_dataset_numpy.shape[1]):#residue index
opo_dataset_numpy[i,j,:] = opo_dataset[i][opo_index[j]]
np.save('dataset/dataset/opo_dataset.npy', opo_dataset_numpy)
print(opo_dataset_numpy.shape)
#计算距残基之间的距离
opo_distance_dataset_numpy = np.zeros((1000, len(opo_index), len(opo_index))) #残基对距离
for i in range(1000):
snapshot = opo_dataset_numpy[i]
k = cdist(snapshot, snapshot, metric='euclidean')
opo_distance_dataset_numpy[i] = k
#原来是:(snapshot No., 残基序号, 残基序号),换成(残基序号,残疾序号,snapshot No)
opo_distance_dataset_numpy = opo_distance_dataset_numpy.swapaxes(0,2)
opo_distance_dataset_numpy = opo_distance_dataset_numpy.reshape(-1, 1000)
np.save('dataset/dataset/opo_distance_dataset.npy', opo_distance_dataset_numpy)
print(opo_distance_dataset_numpy.shape)
#保存成csv吧
index = []
for i in opo_index:
for j in opo_index:
index.append(i+' '+j)
opo_csv = pd.DataFrame(opo_distance_dataset_numpy, index=index, columns=range(1000))
opo_csv.to_csv('dataset/dataset/opo_csv.csv')
opo_csv.head(10)
输出结果如下:

友情提示:
这个太累呀,整完还要写。。。也许会断更。。。。